基因plmyb1r及其在防治荔枝霜疫霉病中的应用
技术领域
1.本发明属于农作物病害绿色防控技术领域,具体涉及基因plmyb1r及其在防治荔枝霜疫霉病中的应用。
背景技术:
2.荔枝霜疫霉(peronophythora litchii chen ex ko et al.)侵染引起的病害为荔枝霜疫病,其寄主植物是荔枝。该病原菌属于鞭毛菌亚门,卵菌纲(oomycetes),霜霉目(peronosporales),霜疫霉科(peronophthoraceae)。由荔枝霜疫霉引起的荔枝霜疫病在荔枝上危害最为严重,且尚未发现有效的抗病荔枝种植品种。目前对荔枝霜疫霉的防治仍然是以农药防治为主,其抗药性风险大并且污染环境,因此研究荔枝霜疫霉的致病机制有非常重要的意义。
3.研究表明,荔枝霜疫霉成功侵染荔枝主要依赖于一系列致病因子。因此,充分挖掘荔枝致病相关基因并开展功能研究,对有效控制荔枝霜疫霉的危害和选育抗病品种具有重要意义。
4.myb蛋白家族存在于所有的真核生物中,其是一类数量繁多,功能多样的转录调节因子,它参与调控生物体的生长发育,生物或非生物诱发的胁迫反应等。
技术实现要素:
5.针对现有技术中存在的缺陷或不足,本发明的首要目的在于提供一种荔枝霜疫霉致病相关蛋白plmyb1r。
6.本发明的另一目的在于提供上述荔枝霜疫霉致病相关蛋白plmyb1r相关的生物材料。
7.本发明的另一目的在于提供上述荔枝霜疫霉致病相关蛋白plmyb1r或生物材料的应用。
8.为了实现上述发明目的,本发明采用以下技术方案:
9.本发明公开了荔枝霜疫霉一个此前未知的蛋白plmyb1r及其编码基因plmyb1r。基因plmyb1r的全长如seq id no.1所示,其cds序列如seq id no.2所示,共编码476个氨基酸,如seq id no.3所示。本发明利用crispr/cas9基因编辑技术构建pbssk::plmyb1r和pyf2.3g-ribo-sgrna1::plmyb1r和pyf2.3g-ribo-sgrna2::plmyb1r敲除载体并通过peg介导的原生质体转化技术敲除plmyb1r基因。最终获得敲除突变体t7、t38、t73(按照转化子验证顺序编号命名),三个突变体在荔枝霜疫霉生长发育过程中存在明显缺陷。致病性测定结果表明,基因plmyb1r的敲除突变体t7、t38、t73对荔枝嫩叶(品种:桂味)的致病力显著降低。上述试验证明。荔枝霜疫霉plmyb1r基因为荔枝霜疫霉的致病相关基因。此外,plmyb1r基因在荔枝霜疫霉中存在两个转录本,我们分别将两个转录本在荔枝霜疫霉中过表达,一共得到三个过表达转化子oe2、oe8和oe10,经致病力测定后发现plmyb1r基因过表达转化子不影响其致病力。
10.一种荔枝霜疫霉致病相关蛋白plmyb1r,其氨基酸序列如seq id no.3所示,或者是如seq id no.3通过一个或多个氨基酸替换、插入、缺失而获得的仍具有相同或相似功能的类似序列所示。
11.上述荔枝霜疫霉致病相关蛋白plmyb1r相关的生物材料,为下述生物材料中的任意一种或多种组合:
12.1)编码所述荔枝霜疫霉致病相关蛋白plmyb1r的核酸分子;
13.2)含有1)中所述核酸分子的表达盒;
14.3)含有1)中所述核酸分子的重组载体,或含有2)中所述表达盒的重组载体;
15.4)含有1)中所述核酸分子的重组微生物,或含有2)中所述表达盒的重组微生物,或含有3)所述重组载体的重组微生物;
16.5)抑制或阻断所述荔枝霜疫霉致病相关蛋白plmyb1r的基因表达的核酸序列;
17.6)利用5)中所述核酸序列制备得到的抑制或阻断所述荔枝霜疫霉致病相关蛋白plmyb1r的基因表达的基因敲除载体;
18.7)利用6)中所述基因敲除载体制备得到的所述荔枝霜疫霉致病相关蛋白plmyb1r的基因缺陷型荔枝霜疫霉。
19.进一步地,1)中所述核酸分子为所述荔枝霜疫霉致病相关蛋白plmyb1r的基因序列,如seq id no:1所示,或者是所述荔枝霜疫霉致病相关蛋白plmyb1r的cds序列,如seq id no:2所示,或者是如seq id no.1通过碱基插入、缺失、或替换而获得的仍具有相同或相似功能的类似序列所示。
20.进一步地,5)中所述核酸序列为plmyb1r基因的反义rna、sirna、shrna或sgrna。
21.更进一步地,所述sgrna如下任意一种序列所示:
22.sgrna1:5'-caactacctgatgagtccgtgaggacgaaacgagtaagctcgtcgtagttgccctaatggatag-3';
23.sgrna2:5'-ctcgttgctgatgagtccgtgaggacgaaacgagtaagctcgtccaacgacagcagaatggatg-3'。
24.上述荔枝霜疫霉致病相关蛋白plmyb1r或生物材料的应用,为下述应用中的任意一种或多种组合:
25.ⅰ
)在调控荔枝霜疫霉致病力中的应用;
26.ⅱ
)在调控荔枝霜疫霉致病相关基因表达水平中的应用;
27.ⅲ
)在调控荔枝霜疫霉生长中的应用;
28.ⅳ
)在调控荔枝霜疫霉卵孢子形成中的应用;
29.ⅴ
)在调控荔枝霜疫霉在胁迫环境下的耐受性中的应用;
30.ⅵ
)在防治荔枝霜疫病中的应用;
31.ⅶ
)作为靶点在设计和筛选抗荔枝霜疫霉药物中的应用。
32.进一步地,
ⅴ
)中所述的胁迫环境包括但不限于细胞壁胁迫、高渗胁迫。
33.更进一步地,所述的细胞壁胁迫为至少25μg
·
ml-1
十二烷基硫酸钠(sds)、至少350μg
·
ml-1
刚果红(cr)或至少100μg
·
ml-1
荧光增白剂(cfw)诱导的细胞壁胁迫。
34.更进一步地,所述的高渗胁迫为至少0.05mol
·
l-1
nacl,至少0.1mol
·
l-1
cacl2或至少0.2mol
·
l-1
山梨醇(sorbitol)诱导的高渗透压胁迫。
35.一种防治由荔枝霜疫霉导致的荔枝霜疫病的方法,通过抑制或阻断所述荔枝霜疫霉致病相关蛋白plmyb1r的基因表达实现。
36.一种抗荔枝霜疫霉药物筛选模型,所述的药物筛选模型为所述荔枝霜疫霉致病相关蛋白plmyb1r的基因缺陷型荔枝霜疫霉。
37.上述抗荔枝霜疫霉药物筛选模型的构建方法,包括如下步骤:
38.(1)根据plmyb1r基因序列利用sgrna网站设计sgrna,将sgrna与pyf2.3g-ribo-sgrna载体进行连接,得到plmyb1r基因敲除质粒pyf2.3g-ribo-sgrna::plmyb1r;
39.或者,根据plmyb1r基因序列上游、下游各约1kb的序列设计左、右同源臂扩增引物,以荔枝霜疫霉基因组dna为模板扩增左右同源臂,与pbssk载体进行连接,得到plmyb1r基因敲除质粒pbssk::plmyb1r;
40.(2)将plmyb1r基因敲除质粒pyf2.3g-ribo-sgrna::plmyb1r或pbssk::plmyb1r导入荔枝霜疫霉野生型菌株的原生质体内,经筛选、验证,获得plmyb1r基因敲除突变体,即为所述的荔枝霜疫霉药物筛选模型。
41.进一步地,步骤(1)中所述的sgrna如下任意一种序列所示:
42.sgrna1:5'-caactacctgatgagtccgtgaggacgaaacgagtaagctcgtcgtagttgccctaatggatag-3';
43.sgrna2:5'-ctcgttgctgatgagtccgtgaggacgaaacgagtaagctcgtccaacgacagcagaatggatg-3'。
44.进一步地,步骤(1)中所述的左、右同源臂扩增引物分别如下所示:
45.左同源臂扩增引物:
46.plmyb1r-left-f:5'-tagaactagtggatcccccgtgctcaatatcttcgtgcgtg-3';
47.plmyb1r-left-r:5'-ttagtctcagaaaattcagcccccaatataagcaagac-3';
48.右同源臂扩增引物:
49.plmyb1r-right-f:5'-agaaaattcagcccccaatataagcaagacatcagtcag-3';
50.plmyb1r-right-r:5'-atgcgtccgagcgaggtagggctgcaggaattcgata-3'。
51.本发明相对于现有技术具有如下的优点及效果:
52.本发明通过试验证明,将基因plmyb1r的上下游序列分别扩增出来,与载体pbssk(南京农业大学植物保护学院卵菌与真菌分子生物学实验室惠赠)相连,同源重组后通过聚乙二醇(polyethylene glycol,peg)介导的原生质体转化技术与crispr/cas9敲除策略,所获得的突变体与野生型相比,生长速率减慢,卵孢子产量显著增多,在多种胁迫环境下敏感性不同,致病性试验表明,基因plmyb1r的缺失显著降低了荔枝霜疫霉的致病力。本发明证实基因plmyb1r是荔枝霜疫霉生长发育、卵孢子的形成和致病性所必需的。我们的研究有助于深入阐明荔枝霜疫霉的致病分子机制,为发现新型药物作用靶标和设计新型高效、低毒安全的杀菌剂提供理论依据。
附图说明
53.图1是荔枝霜疫霉plmyb1r基因同源重组的构建示意图;
54.图2是荔枝霜疫霉plmyb1r基因敲除转化子的pcr扩增结果图;其中,泳道wt:荔枝霜疫霉野生型;泳道ck:经peg转化但未敲除转化子;泳道t7、t38、t73分别表示基因plmyb1r
的三个已敲除转化子;
55.图3是野生型wt、未敲除转化子ck、敲除突变体t7、t38、t73在ca培养基上的生长速率及菌落形态图;其中,a是菌落形态图,b是生长速率统计结果;
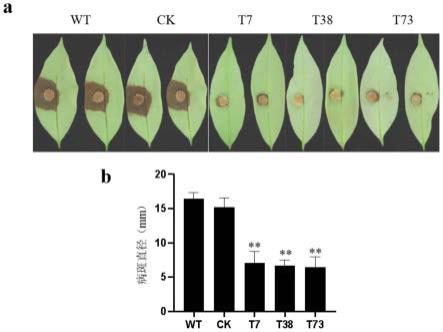
56.图4是基因plmyb1r敲除突变体t7、t38、t73的致病力分析结果;其中,a是侵染病斑图,b是侵染病斑直径统计结果;
57.图5是基因plmyb1r敲除突变体t7、t38、t73对细胞壁胁迫的耐受性结果;其中,a是菌落形态图,b是抑制率统计结果;
58.图6是基因plmyb1r敲除突变体t7、t38、t73对渗透压胁迫的耐受性结果;其中,a是菌落形态图,b是抑制率统计结果;
59.图7是基因plmyb1r敲除突变体t7、t38、t73的卵孢子产量分析结果;其中,a是卵孢子形态图,b是卵孢子产量统计结果。
具体实施方式
60.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
61.下面实施方案中若未注明具体试验条件,则通常按照常规试验条件或按照试剂公司所建议的试验条件。所使用的的材料、试剂等,若无特殊说明,均为从商业途径得到的试剂和材料。
62.实施例1:plmyb1r基因敲除载体的构建
63.试验材料
64.供试菌株、植物与载体:
65.供试菌株为荔枝霜疫霉野生型菌株(peronophythora litchii,wild type,简称wt)为常规的疫霉菌,可通过商业途径或自然界分离获得,大肠杆菌(eschrichia coli)菌株dh5α,可通过商业途径得到;供试接种植物材料是荔枝(桂味)嫩叶(采自华南农业大学园艺实习果园);卵菌敲除及转化载体pyf2-psnls-hspcas9、pyf2.3g-ribo-sgrna(已在文献“yufeng,fang,linkai,et al.efficient genome editing in the oomycetephytophthora sojaeusing crispr/cas9[j].current protocols in microbiology,2017”中公开)、pbssk(已在文献“a crispr/cas9-mediated in situ complementation method for phytophthora sojae mutants[j].molecular plant pathology,2021,22(3)”中公开,以上载体均由南京农业大学植物保护学院卵菌与真菌分子生物学实验室惠赠)。
[0066]
主要供试培养基:
[0067]
胡萝卜榨汁培养基(carrot agar,ca)(1l):胡萝卜300g榨汁,纱布过滤,121℃,20min灭菌。固体培养基加入1.5%(w/v)琼脂粉。
[0068]
lb培养基(1l):5g酵母提取物(yeast extract),10g胰蛋白胨(tryptone),10g氯化钠(nacl),121℃,20min灭菌。固体培养基加入1.5%(w/v)琼脂粉。
[0069]
营养豌豆培养基(nutrition pea broth,npb)(1l):新鲜豌豆120g加水煮20min后过滤。滤液中加入5g山梨醇(d-sorbitol),5g甘露醇(d-mannitol),5g葡萄糖,3g硝酸钾(kno3),2g碳酸钙(caco3),2g酵母提取物,1g磷酸氢二钾(k2hpo
4)
,1g磷酸二氢钾(kh2po4),
0.5g硫酸镁(mgso4),0.1g氯化钙(cacl2),2ml维生素混合液(vitamin stock)和2ml痕量元素(trace elements),加ddh2o定容至1l,121℃,灭菌20min。固体培养基则另加入1.5%(w/v)difco bacto agar。
[0070]
豌豆甘露醇培养基(pea/0.5mol
·
l-1
manitol,pm)(1l):新鲜豌豆120g,加水煮20min后过滤。滤液中加入91g d-mannitol,2g caco3和1.32g cacl2,加ddh2o定容至1l,121℃,灭菌20min。固体培养基则另加入1.5%(w/v)difco bacto agar。
[0071]
皮氏培养基(1l):0.5g磷酸二氢钾,0.25g七水硫酸镁(mgso4·
7h2o),1g l-天冬酰胺(l-asparagine),1mg维生素b1(vitamin b1),0.5g酵母提取物,10mgβ-谷甾醇(β-sitosterol)和5g葡萄糖,加入ddh2o定容至1l,121℃,20min灭菌。固体培养基加入1.5%琼脂粉。
[0072]
crispr/cas9技术相关载体的构建:
[0073]
(1)pyf2.3g-ribo-sgrna::plmyb1r的构建
[0074]
根据设计sgrna网站(http://grna.ctegd.uga.edu/),选择plmyb1r基因上的靶向rna(sgrna),交由生工生物合成后,参照表1配制sgrna退火体系,金属浴37℃,30min。
[0075]
sgrna序列如下:
[0076]
plmyb1r-sgrna1-f:5'-ctagcaactacctgatgagtccgtgaggacgaaacgagtaagctcgtcgtagttgccctaatggatag-3'
[0077]
plmyb1r-sgrna1-r:5'-gttgatggactactcaggcactcctgctttgctcattcgagcagcatcaacgggattacctatccaaa-3'
[0078]
plmyb1r-sgrna2-f:5'-ctagctcgttgctgatgagtccgtgaggacgaaacgagtaagctcgtccaacgacagcagaatggatg-3'
[0079]
plmyb1r-sgrna2-r:5'-gagcaacgactactcaggcactcctgctttgctcattcgagcaggttgctgtcgtcttacctaccaaa-3'。
[0080]
表1 sgrna双链合成体系(30μl)(takara)
[0081][0082]
反应条件为37℃,30min,随后在上述体系内加入4μl 0.5mol
·
l-1
nacl,混匀后于沸水浴煮沸2min,置于室温冷却3~4h,dna片段退火形成双链。
[0083]
plmyb1r基因上的靶向rna(sgrna)退火形成双链,与酶切后pyf2.3g-ribo-sgrna载体(酶切体系见表2)用t4-dna连接酶连接,连接体系如表3,反应条件为16℃,4h,置于冰上冷却,随后进行大肠杆菌转化。
[0084]
表2 pyf2.3g-ribo-sgrna载体双酶切体系(50μl)(neb)
[0085][0086]
表3 pyf2.3g-ribo-sgrna载体与双链sgrna连接反应体系(10μl)(neb)
[0087][0088]
(2)pbssk::plmyb1r载体的构建
[0089]
根据plmyb1r基因上游、下游各约1kb的序列设计左右同源臂和扩增引物(plmyb1r-left-f、plmyb1r-left-r,plmyb1r-right-f、plmyb1r-right-r)。
[0090]
plmyb1r-left-f:5'-tagaactagtggatcccccgtgctcaatatcttcgtgcgtg-3'
[0091]
plmyb1r-left-r:5'-ttagtctcagaaaattcagcccc caatataagcaagac-3'
[0092]
plmyb1r-right-f:5'-agaaaattcagcccc caatataagcaagacatcagtcag-3'
[0093]
plmyb1r-right-r:5'-atgcgtccgagcgaggtagggctgcaggaattcgata-3'。
[0094]
用高保真酶phanta max super-fidelity dna polymerase(南京诺唯赞)进行pcr扩增,以荔枝霜疫霉基因组dna为模板扩增左右同源臂,具体pcr扩增体系如下表4:
[0095]
表4 phanta max高保真酶pcr扩增体系(50μl)
[0096][0097]
扩增程序为:预变性95℃,3min,变性95℃,15s,退火56~72℃,15s,延伸72℃,30s/kb,进行34个循环,最后再延伸5min。目的条带经电泳检测后使用omega公司的琼脂糖
凝胶纯化回收试剂盒进行回收,具体步骤参考试剂盒的说明书。产物检测浓度后用定向无缝克隆试剂盒clonexpress one step cloning kit(南京诺唯赞)与线性化的pbssk载体(酶切体系见表5)连接,连接体系如表6。
[0098]
表5 pbssk酶切反应体系(10μl)
[0099][0100]
表6 pbssk::plmyb1r载体连接反应体系(10μl)
[0101][0102]
将构建好的pyf2.3g-ribo-sgrna1::plmyb1r、pyf2.3g-ribo-sgrna2::plmyb1r、pbssk::plmyb1r、进行大肠杆菌转化。
[0103]
(3)大肠杆菌转化及验证
[0104]
100μl分装的大肠杆菌感受态细胞dh5α冰上冻融,加入连接产物10μl,指尖轻拍混匀,冰上静置30min。42℃水浴热击90s,随后迅速置于冰上2min。650μl lb液体培养基加入管内,37℃,180rpm培养1h。将上述菌液4000rpm离心4min,吸取上清,剩余100μllb培养基悬浮菌体,涂布于含终浓度100μg
·
ml-1
amp的lb固体筛选平板上,37℃培养12~16h。
[0105]
以大肠杆菌单菌落为模板,pyf2.3g-ribo-sgrna载体的验证引物为m13f,rpl41_pseq_f;pbssk载体验证引物为m13f,m13r;其中m13f及m13r为通用引物,引物rpl41_pseq_f的序列如下:rpl41_pseq_f:5'-caagcctcactttctgctgactg-3'。
[0106]
用green taq mix(南京诺唯赞)进行菌落pcr验证,体系如下表7:
[0107]
表7菌落pcr反应体系(20μl)
[0108][0109]
pcr扩增程序为:预变性94℃,5min,变性94℃,30s,退火60℃,30s,延伸72℃,30s/kb,进行34个循环,再延伸7min。扩增产物经凝胶电泳检测,分别选2个扩增条带符合目的片段大小的菌落,用含100μg
·
ml-1
amp的lb液体培养基摇菌送测序。
[0110]
(4)质粒dna的大量提取
[0111]
选择含有目的质粒的大肠杆菌单菌落进行摇菌,单菌落加入至含100μg
·
ml-1
amp的lb液体培养基,37℃,180rpm,培养12h,加入至200ml含amp的lb液体培养基中37℃,180rpm,培养14h。
[0112]
使用tiangen公司的无内毒素质粒大量提取试剂盒(endofree maxi plasmid kit)提取peg介导转化所需的4种质粒(pyf2-nls-hspcas9、pyf2.3g-ribo-sgrna1::plmyb1r、pyf2.3g-ribo-sgrna2::plmyb1r、pbssk::plmyb1r)
[0113]
实施例2:荔枝霜疫霉原生质体的制备及peg介导的转化
[0114]
酶解液的制备:称取0.15g溶壁酶(lysing enzymes,sigma)和0.06g纤维素酶(cellulase,sigma)于灭菌烧杯中,超净台内加入10ml 0.8mol
·
l-1
mannitol,8ml灭菌ddh2o,800μl 0.5mol
·
l-1
kcl,800μl 0.5mol
·
l-1
mes-koh和400μl 0.5mol
·
l-1
cacl2,充分溶解后转移至50ml离心管,待用;
[0115]
w5 solution的制备:称取7.8g glucose,4.6g cacl2,2.25g nacl,0.093g kcl,加入ddh2o定容至250ml,待用;
[0116]
mmg solution的制备:称取甘露醇18.22g、mgcl2·
6h2o 0.76g、mes buffer 2ml,加水定容至250ml,待用;
[0117]
荔枝霜疫霉野生型菌株wt在营养豌豆固体平板培养基(npb固体培养基)上活化。取菌丝块放入锥形瓶中,加入50ml npb液体培养基进行培养,共培养三瓶,于25℃黑暗培养3d,每隔12h摇晃一次。纱布过滤,收集菌丝,用镊子轻轻挤压后加入到含酶解液的50ml离心管中,轻轻混匀后,25℃,40rpm,酶解40~45min。待菌丝酶解后,迅速用包有三层miracloth滤布的50ml烧杯过滤菌丝,并将滤液转移至50ml圆底离心管中,4℃,1500rpm离心3min。弃上清,加入10ml w5 solution重悬浮原生质体,再加入25ml w5 solution,上下颠倒轻轻混匀,4℃,1500rpm离心4min。弃上清,加入7ml w5 solution重悬浮原生质体,置于冰上放置30min。随后4℃,1500rpm离心4min,弃上清,加入6ml的mmg solution重悬浮原生质体,温室放置10min。取6支灭菌50ml离心管置于冰上,向每个离心管中加入4种质粒:pyf2-nls-hspcas9、pyf2.3g-ribo-sgrna1::plmyb1r、pyf2.3g-ribo-sgrna2::plmyb1r、pbssk::plmyb1r,各30μg,得到含原生质体的mmg solution。
[0118]
向每个50ml离心管中加入1ml含原生质体的mmg solution,轻轻摇晃混匀,置于冰
上10min。沿管壁向每个离心管加入580μl 40%聚乙二醇(peg)溶液,连续加入3次。此过程中缓慢旋转离心管,使peg与原生质体混匀,冰上放置20min。豌豆甘露醇培养液(pm)中1000:1加入100mg
·
ml-1
氨苄青霉素制备得到amp pm。在离心管中加入2ml amp pm,轻轻上下颠倒,冰上放置2min;继续在离心管中加入8ml amp pm,并轻轻上下颠倒,冰上放置2min;最后各个离心管中依次加入10ml amp pm,并轻轻上下颠倒,倾斜放置。25℃下黑暗培养14~16h,使原生质体再生。过夜培养后,显微镜下观察原生质体再生情况,随后2000rpm离心5min。各离心管弃上清至剩余5ml液体培养基,悬浮沉淀后加入30ml含30μg
·
ml-1
遗传霉素g418的豌豆甘露醇固体培养基,颠倒混匀后倒入2个9cm灭菌培养皿中,25℃黑暗培养2~3d。挑取单菌落对其进行编号命名,用于鉴定。
[0119]
实施例3:验证和测定
[0120]
(1)plmyb1r基因敲除转化子的验证分析
[0121]
将荔枝霜疫霉野生型wt与转化子用ctab法进行基因组dna提取,以基因组dna为模板,设计基因组中plmyb1r左右同源臂片段外的引物plmyb1rby-f、plmyb1rby-r分别进行常规pcr扩增。通过凝胶电泳检测条带大小,验证plmyb1r是否成功敲除并送测序检测。
[0122]
plmyb1rby-f:5'-agctccagcgcgagagagaac-3'
[0123]
plmyb1rby-r:5'-ggacccacgacagctttcttagc-3'。
[0124]
测序结果证明,peg转化敲除成功,并获得3个plmyb1r基因敲除转化子,依照转化子验证顺序从t1起进行编号,3个敲除成功的突变体分别命名为t7、t38、t73。
[0125]
(2)敲除突变体生长速率的测定
[0126]
荔枝霜疫霉野生型wt菌株、在含g418抗生素培养基生长但敲除不成功的转化子ck、以及plmyb1r基因敲除成功的突变体t7、t38、t73在无抗生素的ca平板转代2次,打孔取菌龄一致的9mm直径的wt,ck和t7、t38、t73菌丝块,接种在15ml等量胡萝卜培养基平板(直径=9cm)中央。设3个重复,25℃黑暗培养5d,测量菌落直径大小并计算生长速率并拍照。实验独立重复三次,用spss软件中的duncan’s multiple range test进行菌株间的差异显著性分析。
[0127]
生长速率(mm/d)=第5天菌落直径/5天。
[0128]
(3)敲除突变体致病力测定
[0129]
荔枝(品种为桂味)嫩叶经ddh2o浸泡后置于湿润滤纸上,取测定步骤(2)中转代培养得到的菌龄一致的9mm直径的wt,ck和t7、t38、t73菌丝块接种,将菌丝生长面覆盖于叶片背面。每个菌株重复接种6片叶龄相近嫩叶,于25℃保湿放置,48h后拍照并测量病斑直径。使用spss软件中的duncan’s multiple range test进行差异显著性分析。
[0130]
(4)敲除突变体对细胞壁胁迫的耐受性测定
[0131]
取测定步骤(2)中转代培养得到的菌龄一致的9mm直径的wt,ck和t7、t38、t73菌丝块接种于定量的皮式培养基中央,作为空白对照。实验组使用分别添加了细胞壁胁迫物质25μg
·
ml-1
十二烷基硫酸钠(sds),350μg
·
ml-1
刚果红(cr),100μg
·
ml-1
荧光增白剂(cfw)的皮式培养基,也用相同的方法接种各敲除突变体菌株的菌丝块。在25℃下黑暗培养7天,测量对照组和实验组的菌落直径并拍照,计算wt、ck、t7、t38、t73在各胁迫因子下的生长抑制率,统计plmyb1r基因敲除突变体的抑制率较wt和ck是否具有显著差异。
[0132]
(5)敲除突变体对高渗压力的耐受性测定
[0133]
本试验通过在皮式固体培养基中添加0.05mol
·
l-1
nacl,0.1mol
·
l-1
cacl2,0.2mol
·
l-1
山梨醇(sorbitol)三种常用的渗透剂,并分别接种测定步骤(2)中转代培养得到的菌龄一致的9mm直径的wt,ck和t7、t38、t73菌丝块。在25℃下黑暗培养7天,测量菌落生长直径和拍照,计算wt,ck,plmyb1r基因敲除突变体在各胁迫因子下的生长抑制率。
[0134]
(6)敲除突变体卵孢子产量测定
[0135]
在经过两次转代培养的wt、ck、t7、t38、t73的菌落边缘用灭菌打孔器取下大小一致的菌碟(d=9mm)并转接于表面覆盖一层hybond n
膜的胡萝卜培养基上,25℃条件下暗培养10天后,揭下滤膜,于接种点附近随机用打孔器打5个菌碟,置于5ml ddh2o中匀浆,吸取1μl于载玻片上统计各个菌株卵孢子数量。
[0136]
实施例4:结果与分析
[0137]
(1)荔枝霜疫霉plmyb1r基因重组片段的构建
[0138]
应用pcr技术分别克隆获得了plmyb1r基因同源臂left、同源臂right片段,多片段连接至线性化pbssk载体,成功得到了pbssk::plmyb1r载体;双链合成获得了sgrna,连接至线性化pyf2.3g-ribo-sgrna载体,成功得到了pyf2.3g-ribo-sgrna1::plmyb1r和pyf2.3g-ribo-sgrna2::plmyb1r载体,敲除示意图如图1。
[0139]
(2)荔枝霜疫霉基因plmyb1r敲除突变体的筛选
[0140]
设计基因plmyb1r左右臂外引物:plmyb1rby-f/r,提取荔枝霜疫霉野生型(wt)、未敲除转化子(ck)与plmyb1r基因敲除突变体的dna,以野生型为对照进行pcr扩增,扩增方法和体系参考实施例1,结果表明,wt与ck扩出约4500bp的片段,t7、t38、t73三个转化子均能扩增出约3000bp的片段,进一步说明t7、t38、t73为基因plmyb1r的敲除转突变体(图2),测序结果证实确已敲除。t7、t38、t73的测序结果如下:
[0141]
ctacaattggtctctccctggtgttatacttcttcacggtacattactagcttccacccccactcactgctcctatttcttcgtacgagacaattcggcagcacatgtgacacaaacactctttcattgttaccacacgtgcatgcactacgttagtctcagaaaattcagcccccaatataagcaagacatcagtcagaaattcgtgagtttcttctgcactcaagtggaacgtacag。
[0142]
(3)基因plmyb1r敲除突变体生长速率的分析
[0143]
与荔枝霜疫霉野生型wt和未敲除转化子ck相比,敲除突变体t7、t38、t73在ca培养基中生长明显减慢,菌丝颜色与野生型相比无显著差异(图3)。
[0144]
(4)基因plmyb1r敲除突变体致病力分析
[0145]
与荔枝霜疫霉野生型wt和未敲除转化子ck相比,敲除突变体t7、t38、t73接种菌饼的病斑直径明显减小。证明基因plmyb1r的缺失导致荔枝霜疫霉的致病力显著降低(图4)。
[0146]
(5)基因plmyb1r敲除突变体对细胞壁胁迫的耐受性分析
[0147]
将敲除突变体t7、t38、t73分别接种至添加25μg
·
ml-1
十二烷基硫酸钠(sds),350μg
·
ml-1
刚果红(cr),100μg
·
ml-1
荧光增白剂(cfw)的皮式培养基,对其菌落直径进行统计并计算抑制率(图5)。结果表明,与野生型wt相比,在含sds胁迫条件下,突变体的耐受性降低,而在cr和cfw的胁迫条件下突变体的耐受性显著升高,说明plmyb1r参与了荔枝霜疫霉对部分胁迫物质引起的细胞壁胁迫的响应。
[0148]
(6)基因plmyb1r敲除突变体对高渗压力的耐受性分析
[0149]
将敲除突变体t7、t38、t73分别接种至添加0.05mol
·
l-1
nacl,0.1mol
·
l-1
cacl2,
0.2mol
·
l-1
山梨醇(sorbitol)的皮氏培养基,对其菌落直径进行统计并计算抑制率(图6)。结果表明,与野生型wt相比,在三种物质的胁迫下突变体的耐受性显著降低,说明plmyb1r参与了荔枝霜疫霉对部分胁迫物质引起的渗透胁迫的响应。
[0150]
(7)基因plmyb1r敲除突变体对卵孢子产量的影响
[0151]
相同处理条件下制备野生型与敲除突变体t7、t38、t73的卵孢子悬浮液,与wt和ck为对照,敲除突变体t7、t38、t73的卵孢子显著升高,证明敲除基因plmyb1r会影响荔枝霜疫霉卵孢子的产量(图7)。
[0152]
实验结果证明,本发明提供的基因可以用于植物病害防治,特别是由荔枝霜疫霉导致的荔枝霜疫病。另外,本发明提供的基因可以作为用于植物病害防治的药物靶标。本领域技术人员可以根据本说明书的指导与启示,开发用于防治植物病害、特别是荔枝霜疫病的药物。
[0153]
上述实例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所做的改变、修饰、替代、组合、简化,均应为等效的替代方案,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。