1.本技术涉及一种cap1帽类似物的合成方法,属于生物化学技术领域。
背景技术:
2.mrna疫苗是近几年兴起一种疫苗形式,尤其是新冠期间表现突出。mrna疫苗具有快速响应、便于大规模制备的优点,同时又没有dna疫苗的遗传变异的风险。非常适合应对突发性的、流传性、季节性的疾病威胁。mrna除了在疫苗领域应用,还可以应用于癌症治疗、遗传性疾病治疗等领域。mrna作为活性药物组分,其核心结构有5’端的帽子结构,5
’‑
utr,基因翻译去、3
’‑
utr和3
’‑
polya尾巴。mrna的帽子结构可以通过转录后使用加帽酶和2-o-甲基转移酶的酶法加帽修饰完成,也可以通过帽类似物共转录加帽方式完成。
3.cap1结构的帽类似物是目前常用的形式,其生产方式是复杂的化学合成。核苷酸上有多个活性基团,因此合成过程中需要到多次的保护和脱保护反应,所以合成产率很低。
技术实现要素:
4.本发明的目的是提供一种cap1帽类似物的合成方法,采用化学合成加酶促反应的方法,简化了传统上的纯化学合成方法。
5.本发明采用的技术手段为:一种cap1帽类似物的合成方法,其步骤包括:通过多步化学反应合成二核苷酸pnpn;二核苷酸pnpn与gdp或者3'-ome-gdp缩合反应生成g-cap结构,即g(5')ppp(5')npn或者(3'-ome)g(5')ppp(5')npn;g(5')ppp(5')npn或者(3'-ome)g(5')ppp(5')npn在加帽酶的催化下生成cap0结构,即m7g(5')ppp(5')npn或者3'-ome-m7g(5')ppp(5')npn;m7g(5')ppp(5')npn或者3'-ome-m7g(5')ppp(5')npn在2-o甲基转移酶的催化下生成cap1帽类似物,即m7g(5')ppp(5')(2'-omen)pn或者(3'-ome-m7g)(5')ppp(5')(2'-omen)pn。
6.优选的,步骤(1)具体为:化合物1、化合物2与吡啶三氟乙酸盐溶于乙腈中搅拌反应,旋蒸旋掉乙腈,加入二氯甲烷,水洗,饱和氯化钠溶液洗涤,柱层析得到产品1;产品1溶解于thf:pyridine:h2o溶剂中,控温至0度,缓慢加入碘单质,室温反应,旋干,加入二氯甲烷,水洗,饱和氯化钠溶液洗涤,旋干,柱层析得到产品2;产品2溶解于chcl3中,控温至0度,缓慢加入三氟乙酸,室温反应,旋干,加入二氯甲烷,水洗,碳酸氢钠溶液洗涤,旋干,柱层析得到化合物3;化合物3 溶解于乙腈中,加入双(2-氰乙基)-n,n-二异丙基亚磷酰胺,及吡啶三氟乙酸盐,室温搅拌过夜,反应完全后加入二氯甲烷及水,分出有机相,碳酸氢钠溶液洗涤,饱和氯化钠溶液洗涤,柱层析得到产品3;
产品3溶解于thf:pyridine:h2o溶剂中,控温至0度,缓慢加入碘单质,室温反应,旋干,加入二氯甲烷,水洗,饱和氯化钠溶液洗涤,旋干,柱层析得到产品4;产品4溶解于thf中,加入浓氨水,室温搅拌过夜,反应完全后旋干得到粗品,柱层析得到二核苷酸pnpn;其中化合物1的结构为,或者化合物1的碱基为尿嘧啶;化合物2的结构为 。
7.优选的,步骤(2)具体为将二核苷酸pnpn溶解于dmf中,加入cdi,加热至50℃反应一段时间,再加入gdp 或者3'-ome-gdp,室温搅拌,反应完后经过离子交换树脂纯化,得到g(5')ppp(5')npn或者(3'-ome)g(5')ppp(5')npn。
8.优选的,步骤(3)和(4)同时进行,即g(5')ppp(5')npn或者(3'-ome)g(5')ppp(5')npn在加帽酶和2-o-甲基转移酶的同时作用下,生成m7g(5')ppp(5')(2'-omen)pn或者(3'-ome-m7g)(5')ppp(5')(2'-omen)pn。
9.优选的,所述加帽酶为牛痘病毒加帽酶,或者加帽酶的甲基转移酶的活性亚基。
10.优选的,所述2-o-甲基转移酶是牛痘病毒来源、黄病毒来源或者冠状病毒来源的2-o-甲基转移酶。
11.本发明采用化学合成加酶促合成的方法合成cap1帽类似物,简化了传统上的纯化学合成方法。起始原料来源更广泛,更便宜,合成步骤更简单,合成得率更高。
附图说明
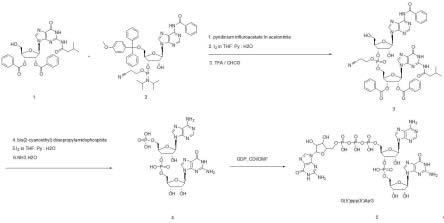
12.图1:化合物5g(5’)ppp(5’)apg的合成路线图。
13.图2:由化合物5g(5’)ppp(5’)apg合成m7g(5')ppp(5')(2'-omen)pg的合成路线图。
14.图3:m7g(5')ppp(5')(2'-omen)pg的结构式。
15.图4:(3'-ome-m7g)(5')ppp(5')(2'-omea)pg的结构式。
具体实施方式
16.实施例1,化学合成g-cap,g(5’)ppp(5’)apg1、化合物3的合成如图1所示,步骤a, 57.5g化合物1加入1.2l 乙腈,溶清后加入85.8g化合物2,57g吡啶三氟乙酸盐,室温搅拌反应过夜,反应完全,旋蒸旋掉乙腈,加入1l 二氯甲烷,水洗一次,饱和食盐水洗涤一次,柱层析得到100g产品1。
17.步骤b,上述100g产品1溶解于1l的thf:pyridine:h2o(4:2:4)溶剂中,控温至0度,缓慢加入3eq的碘单质,室温反应3小时,旋干,加入1000ml二氯甲烷,水洗,饱和氯化钠洗涤,旋干,柱层析得到90g产品2。
18.步骤c,上述90g产品2溶解于1l的chcl3中,控温至0度,缓慢加入3eq的三氟乙酸,室温反应过夜,旋干,加入1000ml二氯甲烷,水洗,5%碳酸氢钠洗涤,旋干,柱层析得到65g化合物3。
19.2、化合物4的合成步骤d,54g 化合物3 溶解于600ml 乙腈中,加入 15g 双(2-氰乙基)-n,n-二异丙基亚磷酰胺,及15g吡啶三氟乙酸盐,室温搅拌过夜,反应完全后加入600ml二氯甲烷及600ml水,分出有机相,5%碳酸氢钠洗涤,饱和氯化钠洗涤,柱层析得到50g产品3。
20.步骤e,上述50g产品3溶解于1l的thf:pyridine:h2o(4:2:4)溶剂中,控温至0度,缓慢加入3eq的碘单质,室温反应3小时,旋干,加入600ml二氯甲烷,水洗,饱和氯化钠洗涤,旋干,柱层析得到40g产品4。
21.步骤f,上述40g产品4溶解于300ml的thf中,加入200ml浓氨水,室温搅拌过夜,反应完全后旋干得到粗品,柱层析得到10g化合物4。
22.3、化合物5的合成步骤g,10g 化合物4溶解于100ml dmf中,加入2g cdi,加热至50度反应2小时,再加入5g gdp,室温搅拌过夜。反应完后经过deae-sephadex离子交换树脂纯化,得到5g 化合物5(g(5’)ppp(5’)apg)。
23.实施例2,化学合成g-cap,(3'-ome)g(5')ppp(5')apg同实施例1相同的合成路线,区别在于3'-ome-gdp替代gdp,得到(3'-ome)g(5')ppp(5')apg。
24.实施例3,酶促合成cap0,m7gpppapg在实施例1中合成的g-cap,g(5’)ppp(5’)apg,取10g,按照下表配置反应体系:原料添加量g(5’)ppp(5’)apg10g氯化镁3.8g二硫苏糖醇3gs-腺苷蛋氨酸32g三羟甲基氨基甲烷2.4g牛痘病毒加帽酶0.1g超纯水至1l在2l反应釜中,控制反应温度为37℃,控制ph为8.0。反应12h后,加入8g edta终止
反应。hplc检测反应完成率为85%。高效液相纯化后纯度为96%,旋转蒸发后,冻干,得到7.5g目标产物m7gpppapg。
25.实施例4,酶促合成cap1,m7gpppampg在实施例3中合成的cap0,m7g(5’)ppp(5’)apg,取5g,按照下表配置反应体系:原料添加量m7g(5’)ppp(5’)apg5g氯化镁2g二硫苏糖醇1.5gs-腺苷蛋氨酸10g三羟甲基氨基甲烷1.2g牛痘病毒2-o-甲基转移酶0.05g超纯水至0.5l在1l反应釜中,控制反应温度为37℃,控制ph为8.0。反应12h后,加入8g edta终止反应。hplc检测反应完成率为92%。高效液相纯化后纯度为98%,旋转蒸发后,冻干,得到4.1g目标产物m7gpppampg。
26.实施例5,一步酶促合成cap1, (3'-ome-m7g)(5')ppp(5')(2'-omea)pg在实施例2中合成的g-cap,(3'-ome)g(5’)ppp(5’)apg,取10g,按照下表配置反应体系:原料添加量(3'-ome)g(5’)ppp(5’)apg10g氯化镁3.8g二硫苏糖醇3gs-腺苷蛋氨酸40g三羟甲基氨基甲烷2.4g非洲猪瘟病毒加帽酶0.1g黄病毒2-o-甲基转移酶0.1g超纯水至1l在2l反应釜中,控制反应温度为37℃,控制ph为8.0。反应12h后,加入8g edta终止反应。hplc检测反应完成率为79%。高效液相纯化后纯度为92%,旋转蒸发后,冻干,得到6.3g目标产物(3'-ome-m7g)(5')ppp(5')(2'-omea)pg。
27.实施例6,合成cap1, m7g(5’)ppp(5’)(2
’‑
omea)pu同实施例1和2一样的合成路线合成g-cap,g(5’)ppp(5’)apu。取10g,按照下表配置反应体系:原料添加量g(5’)ppp(5’)apu10g氯化镁3.8g二硫苏糖醇3gs-腺苷蛋氨酸40g三羟甲基氨基甲烷2.4g
牛痘病毒加帽酶鸟苷转移酶亚基0.05g牛痘病毒2-o-甲基转移酶0.1g超纯水至1l在2l反应釜中,控制反应温度为37℃,控制ph为8.0。反应12h后,加热80℃,30分钟终止反应。hplc检测反应完成率为82%。高效液相纯化后纯度为93%,旋转蒸发后,冻干,得到6.7g目标产物m7g(5')ppp(5')(2'-omea)pu。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。