1.本公开涉及人工智能以及生物医学领域,尤其涉及一种生物标志物确定方法与生物标志物确定装置、计算机可读存储介质及电子设备。
背景技术:
2.在生物医学领域中,生物标志物的确定已经成为了一大重要研究内容。
3.在相关技术中,通常将某一个肿瘤突变频率高的突变基因作为与该肿瘤对应的生物标志物,这导致每次需要确定一个新肿瘤的生物标志物时,都需要首先确定出与新肿瘤对应的突变频率高的突变基因,显然,一方面,这种确定生物标志物的方式不具有通用型,另一方面,若仅根据突变频率确定生物标志物会降低确定出的生物标志物的准确度。
4.鉴于此,本领域亟需开发一种新的生物标志物确定方法及装置。
5.需要说明的是,在上述背景技术部分公开的信息仅用于加强对本公开的背景的理解,因此可以包括不构成对本领域普通技术人员已知的现有技术的信息。
技术实现要素:
6.本公开的目的在于提供一种生物标志物确定方法、生物标志物确定装置、计算机可读存储介质及电子设备,进而至少在一定程度上克服由于相关技术导致的确定出的生物标志物的准确度低的问题。
7.本公开的其他特性和优点将通过下面的详细描述变得显然,或部分地通过本公开的实践而习得。
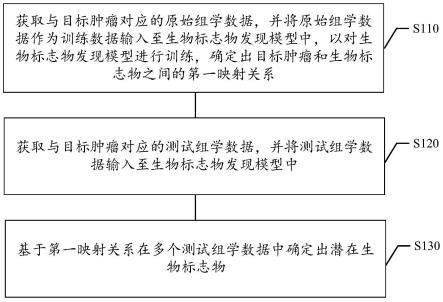
8.根据本发明实施例的第一个方面,提供了一种生物标志物确定方法,所述方法包括:获取与目标肿瘤对应的原始组学数据,并将所述原始组学数据作为训练数据输入至生物标志物发现模型中,以对所述生物标志物发现模型进行训练,确定出所述目标肿瘤和生物标志物之间的第一映射关系;获取与所述目标肿瘤对应的测试组学数据,并将所述测试组学数据输入至所述生物标志物发现模型中;基于所述第一映射关系在所述测试组学数据中确定出潜在生物标志物。
9.在本发明的一种示例性实施例中,所述原始组学数据包括基因突变组学数据、基因甲基组学数据以及蛋白组学数据中的一个或多个;所述获取与目标肿瘤对应的原始组学数据,包括:获取与目标肿瘤对应的训练样本;由每个所述训练样本中与所有基因对应的突变状态组成所述基因突变组学数据;由每个所述训练样本中与所有甲基化基因对应的甲基化值组成所述基因甲基组学数据;由每个所述训练样本中与所有蛋白质对应的相对蛋白丰度组成所述蛋白组学数据。
10.在本发明的一种示例性实施例中,所述由每个所述训练样本中与所述基因对应的突变状态组成所述基因突变组学数据,包括:获取与所述训练样本对应的初始组学数据;其中,所述初始组学数据中包括与不同基因对应的基因位点的位点突变状态;对与同一个所述基因对应的所述基因位点的所述位点突变状态进行求和计算,得到所述基因的基因突变
状态;由每个所述训练样本中与所有所述基因对应的所述基因突变状态组成所述基因突变组学数据。
11.在本发明的一种示例性实施例中,所述初始组学数据还包括所有甲基化基因对应的甲基化值以及与所有蛋白质对应的相对蛋白丰度;所述方法还包括:从所述初始组学数据中获取与所有所述甲基化基因对应的所述甲基化值,并从所述初始组学数据中获取与所有所述蛋白质对应的所述相对蛋白丰度;由每个所述训练样本中与所有甲基化基因对应的甲基化值组成所述基因甲基组学数据;由每个所述训练样本中与所有蛋白质对应的相对蛋白丰度组成所述蛋白组学数据。
12.在本发明的一种示例性实施例中,所述生物标志物发现模型包括一个或多个深度神经网络、一个或多个注意力机制模型以及前向神经网络模型。
13.在本发明的一种示例性实施例中,所述多个深度神经网络包括第一深度神经网络、第二深度神经网络以及第三深度神经网络;所述将多个原始组学数据作为训练数据输入至生物标志物发现模型中,以对所述生物标志物发现模型进行训练,包括:将所述基因突变组学数据输入至所述第一深度神经网络,以将所述基因突变组学数据转换为第一高维特征;将所述基因甲基组学数据输入至所述第二深度神经网络,以将所述基因甲基组学数据转换为第二高维特征;将所述蛋白组学数据输入至所述第三深度神经网络,以将所述蛋白组学数据转换为第三高维特征;对所述第一高维特征、所述第二高维特征以及所述第三高维特征进行两两组合得到多个组合特征,并将所述多个组合特征分别输入至所述多个注意力机制模型中,以得到多个输出特征;将所述多个输出特征输入至所述前向神经网络,以将所述多个输出特征整合为预测组学特征;利用所述预测组学特征,对所述生物标志物发现模型进行训练。
14.在本发明的一种示例性实施例中,所述多个注意力机制模型包括第一注意力机制模型、第二注意力机制模型以及第三注意力机制模型;所述多个输出特征包括第一输出特征、第二输出特征以及第三输出特征;所述对所述第一高维特征、所述第二高维特征以及所述第三高维特征进行两两组合得到多个组合特征,并将所述多个组合特征分别输入至所述多个注意力机制模型中,以得到多个输出特征,包括:获取与所述第一高维特征、所述第二高维特征以及所述第三高维特征分别对应的第一权重、第二权重以及第三权重;利用所述第一权重以及所述第二权重,对所述第一高维特征以及所述第二高维特征进行计算得到第一组合特征,以将所述第一组合特征输入至所述第一注意力机制模型得到所述第一输出特征;利用所述第一权重以及所述第三权重,对所述第一高维特征以及所述第三高维特征进行计算得到第二组合特征,以将所述第二组合特征输入至所述第二注意力机制模型得到所述第二输出特征;利用所述第二权重以及所述第三权重,对所述第二高维特征以及所述第三高维特征进行计算得到第三组合特征,以将所述第三组合特征输入至所述第三注意力机制模型得到所述第三输出特征。
15.在本发明的一种示例性实施例中,所述将所述多个输出特征输入至所述前向神经网络,以将所述多个输出特征整合为预测组学特征,包括:将所述多个输出特征输入至所述前向神经网络,得到待映射特征;获取与所述待映射特征对应的映射公式,并利用所述映射公式对所述待映射特征进行计算,得到预测组学特征。
16.在本发明的一种示例性实施例中,所述方法还包括:获取与所述待映射特征对应
的编码公式,并利用所述编码公式,将所述待映射特征转换为预测样本标签;利用所述预测样本标签,对所述生物标志物发现模型进行训练。
17.在本发明的一种示例性实施例中,所述利用所述预测组学特征,对所述生物标志物发现模型进行训练,包括:若所述原始组学数据中包括已知生物标注物,获取与所述原始组学数据对应的真实样本标签;确定所述真实样本标签与所述预测样本标签之间的第一损失计算关系,并利用所述第一损失计算关系,对所述真实样本标签和所述预测样本标签进行计算得到第一损失计算结果;确定与所述已知生物标志物对应的向量与所述预测组学特征之间的第二损失计算关系,并利用所述第二损失计算关系,对所述向量和所述预测组学特征进行计算得到第二损失计算结果;对所述第一损失计算结果和所述第二损失结算结果进行计算得到第三损失计算结果,根据所述第三损失计算结果,对所述生物标志物发现模型进行训练。
18.在本发明的一种示例性实施例中,所述对所述第一损失计算结果和所述第二损失结算结果进行计算得到第三损失计算结果,包括:确定与所述第一损失结算结果对应的第一损失权重,并确定与所述第二损失计算结果对应的第二损失权重;利用所述第一损失权重和所述第二损失权重,对所述第一损失计算结果和所述第二损失计算结果进行计算得到第三损失计算结果。
19.在本发明的一种示例性实施例中,所述基于所述第一映射关系在所述测试组学数据中确定出潜在生物标志物,包括:基于所述第一映射关系,确定出与所述测试组学数据对应的所述预测组学特征;确定与所述预测组学特征对应的特征元素,对所述特征元素的大小进行比较得到元素比较结果;根据所述元素比较结果,确定出最大特征元素,以将与所述最大特征元素对应的生物标志物确定为潜在生物标志物;其中,所述潜在生物标志物包括所述基因、所述甲基化基因、所述蛋白质中的任意一项。
20.根据本发明实施例的第二个方面,提供一种肿瘤样本筛查方法,所述方法包括:获取与目标肿瘤对应的训练样本,并将与所述训练样本对应的原始组学数据作为训练数据输入至生物标志物发现模型中,以对所述生物标志物发现模型进行训练,确定出所述训练样本与样本标签之间的第二映射关系;获取与所述目标肿瘤对应的测试样本,并将与所述测试样本对应的测试组学数据输入至所述生物标志物发现模型中;基于所述第二映射关系,确定出与所述测试样本对应的所述样本标签,以根据所述样本标签在所述测试样本中筛查出肿瘤样本。
21.在本发明的一种示例性实施例中,所述对所述生物标志物发现模型进行训练,确定出所述训练样本与样本标签之间的第二映射关系,包括:若所述原始组学数据中不包括已知生物标注物,获取与所述原始组学数据对应的真实样本标签;确定所述真实样本标签与所述预测样本标签之间的第一损失计算关系,并利用所述第一损失计算关系,对所述真实样本标签和所述预测样本标签进行计算得到第一损失计算结果;根据所述第一损失计算结果,对所述生物标志物发现模型进行训练,以确定出所述目标肿瘤与样本标签之间的第二映射关系。
22.根据本发明实施例的第三个方面,提供一种生物标志物确定装置,包括:第一训练模块,被配置为获取与目标肿瘤对应的原始组学数据,并将所述原始组学数据作为训练数据输入至生物标志物发现模型中,以对所述生物标志物发现模型进行训练,确定出所述目
标肿瘤和生物标志物之间的第一映射关系;第一输入模块,被配置为获取与所述目标肿瘤对应的测试组学数据,并将所述测试组学数据输入至所述生物标志物发现模型中;预测模块,被配置为基于所述第一映射关系在所述测试组学数据中确定出潜在生物标志物。
23.根据本发明实施例的第四个方面,提供一种肿瘤样本筛查装置,包括:第二训练模块,被配置为获取与目标肿瘤对应的训练样本,并将与所述训练样本对应的原始组学数据作为训练数据输入至生物标志物发现模型中,以对所述生物标志物发现模型进行训练,确定出所述目标肿瘤与样本标签之间的第二映射关系;第二输入模块,被配置为获取与所述目标肿瘤对应的测试样本,并将与所述测试样本对应的测试组学数据输入至所述生物标志物发现模型中;筛查模块,被配置为基于所述第二映射关系,确定出与所述训练样本对应的所述样本标签,以根据所述样本标签在所述测试样本中筛查出肿瘤样本。
24.根据本发明实施例的第五个方面,提供一种电子设备,包括:处理器和存储器;其中,存储器上存储有计算机可读指令,所述计算机可读指令被所述处理器执行时实现上述任意示例性实施例的生物标志物确定方法或上述任意示例性实施例的肿瘤样本筛查方法。
25.根据本发明实施例的第六个方面,提供一种计算机非瞬态可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现上述任意示例性实施例中的生物标志物确定方法或上述任意示例性实施例的肿瘤样本筛查方法。
26.由上述技术方案可知,本发明示例性实施例中的生物标志物确定方法、生物标志物确定装置、计算机存储介质及电子设备至少具备以下优点和积极效果:
27.在本公开的示例性实施例提供的方法及装置中,根据与目标肿瘤具有对应关系的生物标志物发现模型确定出潜在生物标志物,一方面,生物标志物发现模型是将原始组学数据作为训练数据进行训练得到的,并且,原始组学数据与目标肿瘤对应,进而,可以确定出与不同目标肿瘤对应的不同的潜在生物标志物,适用于所有不同的目标肿瘤;另一方面,避免了现有技术中,将突变频率高的突变基因作为生物标志物的情况发生,提高了确定出的潜在生物标志物的准确度。
28.应当理解的是,以上的一般描述和后文的细节描述仅是示例性和解释性的,并不能限制本公开。
附图说明
29.此处的附图被并入说明书中并构成本说明书的一部分,示出了符合本公开的实施例,并与说明书一起用于解释本公开的原理。显而易见地,下面描述中的附图仅仅是本公开的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
30.图1示意性示出本公开实施例中生物标志物确定的流程示意图;
31.图2示意性示出了生物标志物确定方法中获取与目标肿瘤对应的原始组学数据的流程示意图;
32.图3示意性示出了生物标志物确定方法中确定基因突变组学数据的流程示意图;
33.图4示意性示出了生物标志物确定方法中确定基因甲基组学数据以及蛋白组学数据的流程示意图;
34.图5示意性示出了生物标志物确定方法中生物标志物发现模型的结构示意图;
35.图6示意性示出了生物标志物确定方法中对生物标志物发现模型进行训练的流程示意图;
36.图7示意性示出了生物标志物确定方法中得到多个输出特征的流程示意图;
37.图8示意性示出了生物标志物确定方法中生物标志发现模型的模型结构图;
38.图9示意性示出了生物标志物确定方法中将多个输出特征整合为预测组学特征的流程示意图;
39.图10示意性示出了生物标志物确定方法中对生物标志物发现模型进行训练的流程示意图;
40.图11示意性示出了生物标志物确定方法中对生物标志物发现模型进行训练的流程示意图;
41.图12示意性示出了生物标志物确定方法中对第一损失计算结果和第二损失计算结果进行计算得到第三损失计算结果的流程示意图;
42.图13示意性示出了生物标志物确定方法中确定出潜在生物标志物的流程示意图;
43.图14示意性示出本公开实施例中肿瘤样本筛查的流程示意图;
44.图15示意性示出了肿瘤样本筛查方法中对生物标志物发现模型进行训练的流程示意图;
45.图16示意性示出本公开实施例中一种生物标志物确定的装置结构示意图;
46.图17示意性示出本公开实施例中一种肿瘤样本筛查的装置结构示意图;
47.图18示意性示出本公开实施例中一种用于生物标志物确定方法或肿瘤样本筛查方法的电子设备;
48.图19示意性示出本公开实施例中一种用于生物标志物确定方法或肿瘤样本筛查方法的计算机非瞬态可读存储介质。
具体实施方式
49.现在将参考附图更全面地描述示例实施方式。然而,示例实施方式能够以多种形式实施,且不应被理解为限于在此阐述的范例;相反,提供这些实施方式使得本公开将更加全面和完整,并将示例实施方式的构思全面地传达给本领域的技术人员。所描述的特征、结构或特性可以以任何合适的方式结合在一个或更多实施方式中。在下面的描述中,提供许多具体细节从而给出对本公开的实施方式的充分理解。然而,本领域技术人员将意识到,可以实践本公开的技术方案而省略所述特定细节中的一个或更多,或者可以采用其它的方法、组元、装置、步骤等。在其它情况下,不详细示出或描述公知技术方案以避免喧宾夺主而使得本公开的各方面变得模糊。
50.本说明书中使用用语“一个”、“一”、“该”和“所述”用以表示存在一个或多个要素/组成部分/等;用语“包括”和“具有”用以表示开放式的包括在内的意思并且是指除了列出的要素/组成部分/等之外还可存在另外的要素/组成部分/等;用语“第一”和“第二”等仅作为标记使用,不是对其对象的数量限制。
51.此外,附图仅为本公开的示意性图解,并非一定是按比例绘制。图中相同的附图标记表示相同或类似的部分,因而将省略对它们的重复描述。附图中所示的一些方框图是功能实体,不一定必须与物理或逻辑上独立的实体相对应。
52.针对相关技术中存在的问题,本公开提出了一种生物标志物确定方法。图1示出了生物标志物确定方法的流程示意图,如图1所示,生物标志物确定方法至少包括以下步骤:
53.步骤s110.获取与目标肿瘤对应的原始组学数据,并将原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,确定出目标肿瘤和生物标志物之间的第一映射关系。
54.步骤s120.获取与目标肿瘤对应的测试组学数据,并将测试组学数据输入至生物标志物发现模型中。
55.步骤s130.基于第一映射关系在多个测试组学数据中确定出潜在生物标志物。
56.在本公开的示例性实施例提供的方法及装置中,根据与目标肿瘤具有对应关系的生物标志物发现模型确定出潜在生物标志物,一方面,生物标志物发现模型是将原始组学数据作为训练数据进行训练得到的,并且,原始组学数据与目标肿瘤对应,进而,可以确定出与不同目标肿瘤对应的不同的潜在生物标志物,适用于所有不同的目标肿瘤;另一方面,避免了现有技术中,将突变频率高的突变基因作为生物标志物的情况发生,提高了确定出的潜在生物标志物的准确度。
57.下面对生物标志物确定方法的各个步骤进行详细说明。
58.在步骤s110中,获取与目标肿瘤对应的原始组学数据,并将原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,确定出目标肿瘤和生物标志物之间的第一映射关系。
59.在本公开的示例性实施例中,目标肿瘤可以是任何一种肿瘤,具体地,目标肿瘤可以是肝癌,可以是乳腺癌,还可以是肺癌,本示例性实施例对此不做特殊限定,值得说明的是,目标肿瘤还可以是两种相关的肿瘤,具体地,目标肿瘤可以是妇科疾病中的乳腺癌和卵巢癌,目标肿瘤还可以是妇科疾病中的卵巢癌和子宫内膜癌,本示例性实施例对此不做特殊限定。
60.原始组学数据与目标肿瘤对应,并且原始组学数据指的是组学在生物医学等领域的研究应用中所收集到的数据,通常情况下,原始组学数据可以从癌症基因组图谱数据库上下载得到。
61.生物标志物指的是一种用于标记器官、组织、细胞以及亚细胞所具有的功能发生改变或可能发生改变的生化指标。生物标志物发现模型用于发现与目标肿瘤具有第一映射关系的潜在生物标志物,并且,在创建生物标志物发现模型时,首先需要将原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练。
62.之所以可以将原始组学数据作为训练数据,用于对生物标志物发现模型进行训练,是因为原始组学数据可以是与正常样本对应的数据,也可以是是与确定患有目标肿瘤的肿瘤样本对应的数据,还可以是与目标肿瘤具有映射关系的已知生物标志物对应的数据,因此,通过将原始组学数据作为训练数据输入至生物标志物发现模型,可以确定出目标肿瘤与生物标志物之间的第一映射关系,进而在对生物标志物发现模型训练完成后,生物标志物发现模型就可以用于确定与测试组学数据具有第一映射关系的潜在生物标志物。
63.举例而言,目标肿瘤为子宫癌,从癌症基因组图谱数据库上可以获取到与子宫癌对应的原始组学数据,基于此,将原始组学数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,在训练完成后,确定出了子宫癌与生物标志物之间的映射关系。
64.在可选的实施例中,图2示出了生物标志物确定方法中获取与目标肿瘤对应的原始组学数据的流程示意图,原始组学数据包括基因突变组学数据、基因甲基组学数据以及蛋白组学数据中的一个或多个,如图2所示,该方法至少包括以下步骤:在步骤s210中,获取与目标肿瘤对应的训练样本。
65.其中,原始组学数据具体可以包括以下三种组学数据中的一个或多个,具体地,三种组学数据包括基因突变组学数据、基因甲基组学数据以及蛋白组学数据,基因突变组学数据指的是描述基因是否发生突变的数据,基因甲基组学数据描述的是基因甲基化程度的数据,蛋白组学数据描述的是蛋白质表达的水平。值得说明的是,原始组学数据还可以是除了上述三种组学数据之外的其他组学数据,本示例性实施例对此不做特殊限定。
66.具体地,在获取基因突变组学数据、基因甲基组学数据以及蛋白组学数据时,首先需要获取到与目标肿瘤对应的训练样本,值得说明的是,这些样本可以是正常样本(与未患目标肿瘤的患者对应的样本),这些样本也可以是与已患目标肿瘤的患者对应的肿瘤样本。
67.在获取到训练样本之后,就可以获取到与训练样本对应的原始组学数据。
68.举例而言,获取到的与子宫癌对应的训练样本包括s001、s002、s003、s004以及s005,基于此,分别获取与训练样本s001、s002、s003、s004以及s005对应的原始组学数据。
69.在步骤s220中,由每个训练样本中与所有基因对应的突变状态组成基因突变组学数据。
70.其中,突变状态用以表示该基因是否已经发生突变。
71.举例而言,表1为基因突变组学数据。
72.表1基因突变组学数据
[0073] s001s002s003s004s005b100101b200100b300010b400010b501010
[0074]
其中,如表1所示,s001、s002、s003、s004以及s005为训练样本,b1、b2、b3、b4以及b5为基因,基于此,基因组学数据具体包括(0 0 0 0 0)、(0 0 0 0 1)、(1 1 0 0 0)、(0 0 1 1 1)以及(1 0 0 0 0)。
[0075]
在步骤s230中,由每个训练样本中与所有甲基化基因对应的甲基化值组成基因甲基组学数据。
[0076]
其中,由每个训练样本中与所有甲基化基因对应的甲基化值组成原始组学数据中的基因甲基组学数据,并且基因甲基组学数据的取值范围为[0 1],当基因甲基组学数据的取值属于区间[0.6 1]时,表示该甲基化基因完全甲基化,当基因甲基组学数据的取值范围属于区间[0 0.2]时,表示该甲基化基因完全未甲基化,当基因甲基组学数据的取值范围在区间(0.2 0.6)时,表示该甲基化基因部分甲基化。
[0077]
值得说明的是,通常而言,甲基化值是根据甲基化基因的甲基化信号强度以及甲基化基因的未甲基化信号强度计算得到的,并且在得到之后将其存储在癌症基因组图谱数据库中。
[0078]
具体地,甲基化值的计算公式如公式(1)所示。
[0079]
β=m/(m u)
ꢀꢀ
(1)
[0080]
其中,β为甲基化值,m为甲基化基因的甲基化信号强度,u为甲基化基因的未甲基化信号强度。
[0081]
举例而言,表2为基因甲基组学数据。
[0082]
表2基因甲基组学数据
[0083] s001s002s003s004s005a10.020.040.040.040.04a20.030.070.070.060.06a30.010.020.020.020.02a40.030.040.040.050.03a50.040.050.050.060.05
[0084]
其中,如表2所示,训练样本包括s001、s002、s003、s004以及s005,甲基化基因包括a1、a2、a2、a4以及a5,基于此,基因甲基组学数据包括(0.02 0.03 0.01 0.03 0.04)、(0.04 0.07 0.02 0.04 0.05)、(0.04 0.07 0.02 0.04 0.05)、(0.04 0.06 0.02 0.05 0.06)以及(0.04 0.06 0.02 0.03 0.05)。
[0085]
在步骤s240中,由每个训练样本中与所有蛋白质对应的相对蛋白丰度组成蛋白组学数据。
[0086]
其中,蛋白组学数据是由与所有蛋白质对应的相对蛋白丰度组成的,相对蛋白丰度表示的是蛋白质的表达水平,若相对蛋白丰度小于0,则表示蛋白质表达水平低,若相对蛋白丰度大于0,则表示蛋白质表达水平高。
[0087]
举例而言,表3为蛋白组学数据。
[0088]
表3蛋白组学数据
[0089] s001s002s003s004s005c1-1.18-0.685-0.5282.35-1.67c2-0.863-1.07-1.322.82-1.19c3-0.802-0.6840.435-1.47-0.443c40.2220.984nana0.243c50.2560.135-0.240.154-0.0993
[0090]
其中,s001、s002、s003、s004以及s005为训练样本,c1、c2、c3、c4以及c5为蛋白质,表3中的数值表示某一个训练样本中某一个蛋白质的相对蛋白丰度,na表示不存在对应的相对蛋白丰度。
[0091]
基于此,(-1.18
ꢀ‑
0.863
ꢀ‑
0.802 0.222 0.256)、(-0.685
ꢀ‑
1.07
ꢀ‑
0.684 0.984 0.135)、(-0.528
ꢀ‑
1.23 0.435 na
ꢀ‑
0.24)、(2.35 2.82
ꢀ‑
1.47 na 0.154)以及(-1.67
ꢀ‑
1.19
ꢀ‑
0.443 0.243
ꢀ‑
0.0993)为蛋白组学数据。
[0092]
在本示例性实施例中,一方面,获取到目标肿瘤的训练样本之后,可以获取与训练样本对应的原始组学数据,这为后续利用原始组学数据对生物标志物发现模型进行训练奠定了基础;另一方面,原始组学数据可以是基因突变组学数据、基因甲基组学数据以及蛋白组学数据中的一个或多个,这使得后续训练完成的生物标志物发现模型可以适用于上述三
个组学数据中的一个或多个,扩大了生物标志物发现模型适用的场景。
[0093]
在可选的实施例中,图3示出了生物标志物确定方法中确定基因突变组学数据的流程示意图,如图3所示,该方法至少包括以下步骤:在步骤s310中,获取与训练样本对应的初始组学数据;其中,初始组学数据中包括与不同基因对应的基因位点的位点突变状态。
[0094]
其中,在获取到训练样本之后,可以获取与该训练样本对应的初始组学数据,初始组学数据包括与不同基因对应的基因位点的位点突变状态,位点突变状态可以描述与基因对应的基因位点是否发生突变。
[0095]
举例而言,获取到的与子宫癌对应的训练样本包括s001、s002、s003、s004以及s005,存在两个基因,分别为a1bg和a1cf,还存在与基因a1bg对应的两个基因位点,分别为298以及181,还存在与基因a1cf对应的两个基因位点,分别为487和236
[0096]
基于此,可以得到如表4所示的基因位点的位点突变状态。
[0097] s001s002s003s004s005a1bg_p.e298k00101a1bg_p.s181k00100a1cf_p.f487l00010a1cf_p.s236y00010
[0098]
在步骤s320中,对与同一个基因对应的基因位点的位点突变状态进行求和计算,得到基因的基因突变状态。
[0099]
对与同一个基因对应的基因位点的位点突变状态进行相加,得到的结果即为该基因的基因突变状态。
[0100]
举例而言,如表4所示,存在两个基因,分别为a1bg和a1cf,还存在与基因a1bg对应的两个基因位点,分别为298以及181,还存在与基因a1cf对应的两个基因位点,分别为487和236。
[0101]
由于基因的突变状态是与该基因对应的基因位点的位点突变状态进行计算得到的,因此,与训练样本s001对应的基因a1bg的突变状态为0 0,即与训练样本s001对应的基因a1bg的突变状态为0,与训练样本s001对应的基因a1cf的突变状态为0 0,即与训练样本s001对应的基因a1cf的突变状态为0;与训练样本s002对应的基因a1bg的突变状态为0 0,即与训练样本s002对应的基因a1bg的突变状态为0,与训练样本s002对应的基因a1cf的突变状态为0 0,即与训练样本s002对应的基因a1cf的突变状态为0;与训练样本s003对应的基因a1bg的突变状态为1 1,即与训练样本s003对应的基因a1bg的突变状态为2,与训练样本s003对应的基因a1cf的突变状态为0 0,即与训练样本s003对应的基因a1cf的突变状态为0;与训练样本s004对应的基因a1bg的突变状态为0 0,即与训练样本s004对应的基因a1bg的突变状态为0,与训练样本s004对应的基因a1cf的突变状态为1 1,即与训练样本s004对应的基因a1cf的突变状态为2;与训练样本s005对应的基因a1bg的突变状态为1 0,即与训练样本s005对应的基因a1bg的突变状态为1,与训练样本s005对应的基因a1cf的突变状态为0 0,即与训练样本s005对应的基因a1cf的突变状态为0。
[0102]
在步骤s330中,由每个训练样本中与所有基因对应的基因突变状态组成基因突变组学数据。
[0103]
其中,在确定出与每个基因对应的基因突变状态之后,由每个训练样本中与所有
基因对应的基因突变状态组成基因突变组学数据。
[0104]
举例而言,根据表4得到的基因突变组学数据为(0 0)、(0 0)、(2 0)、(0 2)以及(1 0)。
[0105]
在本示例性实施例中,对基因位点的位点突变状态进行求和得到与该基因位点对应的基因的突变状态,完善了确定基因的突变状态的逻辑,提高了基因突变组学数据所代表的特征的准确度。
[0106]
在可选的实施例中,图4示出了生物标志物确定方法中确定基因甲基组学数据以及蛋白组学数据的流程示意图,如图4所示,该方法至少包括以下步骤:在步骤s410中,从初始组学数据中获取与所有甲基化基因对应的甲基化值,并从初始组学数据中获取与所有蛋白质对应的相对蛋白丰度。
[0107]
其中,初始组学时候中还包括与所有甲基化基因对应的甲基化值以及与所有蛋白质对应的相对蛋白丰度。
[0108]
甲基化值指的是描述基因甲基化程度的数值,相对蛋白丰度指的是蛋白质相对于参考丰度的表达水平,参考丰度是在多次实验过程中确定处于用于作为参考的值。
[0109]
举例而言,在获取到初始组学数据之后,可以直接从初始组学数据中确定出与所有甲基化基因对应的甲基化值,还可以从初始组学数据中确定出与所有蛋白质对应的相对蛋白丰度。
[0110]
在步骤s420中,由每个训练样本中与所有甲基化基因对应的甲基化值组成基因甲基组学数据。
[0111]
其中,从初始组学数据中确定出与所有甲基化基因对应的甲基化值之后,可以确定出与每个训练样本对应的所有甲基化基因的甲基化值,进而组成基因甲基组学数据。
[0112]
举例而言,从初始组学数据中可以确定出如表2所示的,与所有甲基化基因对应的甲基化值。
[0113]
基于此,确定出的基因甲基组学数据为(0.02 0.03 0.01 0.03 0.04)、(0.04 0.07 0.02 0.04 0.05)、(0.04 0.07 0.02 0.04 0.05)、(0.04 0.06 0.02 0.05 0.06)以及(0.04 0.06 0.02 0.03 0.05)。
[0114]
在步骤s430中,由每个训练样本中与所有蛋白质对应的相对蛋白丰度组成蛋白组学数据。
[0115]
其中,从初始组学数据中确定出与所有蛋白质对应的相对蛋白丰度之后,可以确定出与每个训练样本对应的所有蛋白质的相对蛋白丰度,进而组成蛋白组学数据。
[0116]
举例而言,从初始组学数据中可以确定出如表3所示的,与所有蛋白质对应的先对蛋白丰度。
[0117]
基于此,确定出的蛋白组学数据为(-1.18
ꢀ‑
0.863
ꢀ‑
0.802 0.222 0.256)、(-0.685
ꢀ‑
1.07
ꢀ‑
0.684 0.984 0.135)、(-0.528
ꢀ‑
1.23 0.435 na
ꢀ‑
0.24)、(2.35 2.82
ꢀ‑
1.47 na 0.154)以及(-1.67
ꢀ‑
1.19
ꢀ‑
0.443 0.243
ꢀ‑
0.0993)。
[0118]
在本示例性实施例中,基因甲基组学数据以及蛋白组学数据是根据初始组学数获取到的,为后续对生物标志物发现模型的训练奠定了基础。
[0119]
在可选的实施例中,生物标志物发现模型包括一个或多个深度神经网络、一个或多个注意力机制模型以及前向神经网络模型。
[0120]
其中,具体地,生物标志物发现模型包括一个或多个深度神经网络、一个或多个注意力机制模型以及前向神经网络,具体地,深度神经网络的个数与原始组学数据的个数一致,注意力机制的个数与原始组学数据的个数一致。
[0121]
深度神经网络可以是一种多层加权组合的神经网络,也可以是多层卷积的神经网络,还可以是任何一种深度神经网络,本示例性实施例对此不做特殊限定。注意力机制模型指的是一种利用人类的视觉注意力衍生而成的深度学习模型,前向神经网络指的是一种数据只会向前传递的深度神经网络。
[0122]
举例而言,图5示意性示出了生物标志物发现模型的结构示意图,如图5所示,其中,特征510为原始组学数据,具体地,特征510包括特征511基因突变组学数据、特征512基因甲基组学数据以及特征513蛋白组学数据,模型521、模型522以及模型523为与特征511、特征512以及特征513分别对应的深度神经网络,特征531为特征511经过深度神经网络521后得到的高维特征,特征532为特征512经过深度神经网络522后得到的高维特征,特征533为特征513经过深度神经网络523后得到的高维特征。
[0123]
模型541、模型542以及模型543为多个注意力机制模型,其中,输入注意力机制模型541的输入特征为高维特征531和高维特征532,输入注意力机制模型542的输入特征为高维特征531和高维特征533,输入注意力机制模型543的输入特征为高维特征532和高维特征533,模型550为前向深度神经网络,结果560为将原始组学数据输入至生物标志物发现模型后得到的输出结果。
[0124]
在可选的实施例中,生物标志物发现模型中包括一个或多个深度神经网络以及一个或多个注意力机制模型,进而通过深度神经网络可以刻画出每个原始组学数据内数据之间的相关性,通过注意力机制模型可以刻画出任意两个组学数据之间的相关性,这避免了现有技术不考虑组学数据内部以及组学数据之间的相关性的情况发生,进而提高了确定出的潜在生物标志物的准确度。
[0125]
在步骤s120中,获取与目标肿瘤对应的测试组学数据,并将测试组学数据输入至生物标志物发现模型中。
[0126]
在本公开的示例性实施例中,测试组学数据指的是用于输入至训练好的生物标志物发现模型中,以发现与目标肿瘤对应的潜在生物标志物的数据。
[0127]
举例而言,获取到与子宫癌对应的测试组学数据,并将该测试组学数据输入至生物标志物发现模型中。
[0128]
在可选的实施例中,图6示出了生物标志物确定方法中对生物标志物发现模型进行训练的流程示意图,多个深度神经网络包括第一深度神经网络、第二深度神经网络以及第三深度神经网络,如图5所示,该方法至少包括以下步骤:在步骤s610中,将基因突变组学数据输入至第一深度神经网络,以将基因突变组学数据转换为第一高维特征。
[0129]
其中,具体地,多个深度神经网络可以包括第一深度神经网络、第二深度神经网络以及第三深度神经网络,并且,基因突变组学数据是输入至第一深度神经网络之中的,所得到的输出结果为第一高维特征,即通过第一深度神经网络将基因突变组学数据从低维特征转换为高维特征。
[0130]
举例而言,如图5所示,数据511为基因突变组学数据,模型521为第一深度神经网络,特征531为第一高维特征。
[0131]
在步骤s620中,将基因甲基组学数据输入至第二深度神经网络,以将基因甲基组学数据转换为第二高维特征。
[0132]
其中,基因甲基组学数据是输入至第二深度神经网络之中的,所得到的输出结果为第二高维特征,即通过第二深度神经网络将基因甲基组学数据从低维特征转换为了高维特征。
[0133]
举例而言,如图5所示,数据512为基因甲基组学数据,模型522为第二深度神经网络,特征532为第二高维特征。
[0134]
在步骤s630中,将蛋白组学数据输入至第三深度神经网络,以将蛋白组学数据转换为第三高维特征。
[0135]
其中,蛋白组学数据是输入至第三深度神经网络之中的,所得到的输出结果为第三高维特征,即通过第三深度神经网络将蛋白组学数据从低维特征转换为高维特征。
[0136]
举例而言,如图5所示,数据513为蛋白组学数据,模型523为第三深度神经网络,特征533为第三高维特征。
[0137]
在步骤s640中,对第一高维特征、第二高维特征以及第三高维特征进行两两组合得到多个组合特征,并将多个组合特征分别输入至多个注意力机制模型中,以得到多个输出特征。
[0138]
其中,在得到第一高维特征、第二高维特征以及第三高维特征之后,可以将第一高维特征和第二高维特征进行组合得到一个组合特征,还可以将第一高维特征和第三高维特征进行组合得到另一个组合特征,还可以将第二高维特征和第三高维特征进行组合得到另一个组合特征,在得到上述三个组合特征之后,将三个组合特征分别输入至多个注意力机制模型中,从多个注意力机制模型中分别输出的特征即为多个输出特征。
[0139]
值得说明的是,以上对第一高维特征、第二高维特征以及第三高维特征进行两两组合的过程区别与对特征融合的过程,因此,组合特征并不是对第一高维特征、第二高维特征以及第三高维特征进行两两融合后得到的特征。
[0140]
举例而言,如图5所述,特征531即为第一高维特征,特征532即为第二高维特征,特征533即为第三高维特征,将第一高维特征和第二高维特征进行组合后输入至注意力机制模型541中,将第一高维特征531和第三高维特征533进行组合输入至注意力机制模型542中,将第二高维特征532和第三高维特征533进行组合输入至注意力机制模型543中,进而从注意力机制模型541、注意力机制模型542以及注意力机制模型543中分别输出的特征即为输出特征。
[0141]
在步骤s650中,将多个输出特征输入至前向神经网络,以将多个输出特征整合为预测组学特征。
[0142]
其中,将多个输出特征输入至前向神经网络,以对多个输出特征进行整合得到预测组学特征z。
[0143]
举例而言,如图5所示,将注意力机制模型541、注意力机制模型542以及注意力机制模型543分别输出的输出特征输入至前向神经网络550中,前向神经网络550输出的结果即为预测组学特征。
[0144]
在步骤s660中,利用预测组学特征,对生物标志物发现模型进行训练。
[0145]
其中,预测组学特征是利用生物标志物模型得到的预测结果,由于训练数据为已
知其准确结果的数据,进而将预测结果与准确结果进行损失计算,即可对生物标志物发现模型进行训练,直至训练结束,通常而言,训练结束的条件为训练次数达到预设次数,或对预测结果与准确结果进行损失计算得到的计算结果小于预设值。
[0146]
在本示例性实施例中,将基因突变组学数据、基因甲基组学数据以及蛋白组学数据作为训练数据分别输入至第一深度神经网络、第二深度神经网络以及第三深度神经网络当中,进而可以利用深度神经网络捕捉基因突变组学数据内的相关性、基因甲基组学数据内的相关性以及蛋白组学数据内的相关性,避免了现有技术未考虑单个组学数据内的相关性的情况发生,有助于提高生物标志物发现模型的训练准确度,进而提高后续确定出的潜在生物标志物的准确度。
[0147]
在可选的实施例中,图7示出了生物标志物确定方法中得到多个输出特征的流程示意图,多个注意力机制模型包括第一注意力机制模型、第二注意力机制模型以及第三注意力机制模型;多个输出特征包括第一输出特征、第二输出特征以及第三输出特征,如图7所示,该方法至少包括以下步骤:在步骤s710中,获取与第一高维特征、第二高维特征以及第三高维特征分别对应的第一权重、第二权重以及第三权重。
[0148]
其中,多个注意力机制模型包括第一注意力机制模型、第二注意力机制模型以及第三注意力机制模型。
[0149]
第一权重指的是与第一高维特征对应的权重,第二权重指的是与第二高维特征对应的权重,第三权重指的是与第三高维特征对应的权重。
[0150]
举例而言,获取与第一高维特征对应的第一权重为ε1,与第二高维特征对应的第二权重为ε2,与第三高维特征对应的第三权重为ε3。
[0151]
在步骤s720中,利用第一权重以及第二权重,对第一高维特征以及第二高维特进行计算得到第一组合特征,以将第一组合特征输入至第一注意力机制模型得到第一输出特征。
[0152]
其中,第一组合特征即为通过第一权重和第二权重,对第一高维特征和第二高维特征进行计算后得到的特征,并且在得到第一组合特征之后,将第一组合特征输入至第一注意力机制模型中,以得到第一输出特征。
[0153]
举例而言,如图5所示,特征531为第一高维特征h1,特征532为第二高维特征h2,并且,与第一高维特征对应的第一权重为ε1,与第二高维特征对应的第二权重为ε2,进而第一组合特征可以是ε1
×
h1 ε2
×
h2。
[0154]
基于此,将第一组合特征输入至第一注意力机制模型541中,以得到第一输出特征。
[0155]
在步骤s730中,利用第一权重以及第三权重,对第一高维特征以及第三高维特征进行计算得到第二组合特征,以将第二组合特征输入至第二注意力机制模型得到第二输出特征。
[0156]
其中,第二组合特征即为通过第一权重和第三权重,对第一高维特征和第三高维特征进行计算后得到的特征,并且在得到第二组合特征之后,将第二组合特征输入至第二注意力机制模型中,以得到第二输出特征。
[0157]
举例而言,如图5所示,特征531为第一高维特征h1,特征533为第三高维特征h3,并且,与第一高维特征对应的第一权重为ε1,与第三高维特征对应的第三权重为ε3,进而第二
组合特征可以是ε1
×
h1 ε3
×
h3。
[0158]
基于此,将第二组合特征输入至第二注意力机制模型542中,以得到第二输出特征。
[0159]
在步骤s740中,利用第二权重以及第三权重,对第二高维特征以及第三高维特征进行计算得到第三组合特征,以将第三组合特征输入至第三注意力机制模型得到第三输出特征。
[0160]
其中,第三组合特征即为通过第二权重和第三权重,对第二高维特征和第三高维特征进行计算后得到的特征,并且在得到第三组合特征之后,将第三组合特征输入至第三注意力机制模型中,以得到第三输出特征。
[0161]
举例而言,如图5所示,特征532为第二高维特征h2,特征533为第三高维特征h3,并且,与第二高维特征对应的第二权重为ε2,与第三高维特征对应的第三权重为ε3,进而第三组合特征可以是ε2
×
h2 ε3
×
h3。
[0162]
基于此,将第三组合特征输入至第三注意力机制模型543中,以得到第三输出特征。
[0163]
图8示意性示出了生物标志发现模型的模型结构图,如图8所示,其中,数据811为基因突变组学数据,数据812为基因甲基组学数据,数据813为蛋白组学数据,模型821为第一深度神经网络,模型822为第二深度神经网络,模型823为第三深度神经网络,特征831为第一高维特征,特征832为第二高维特征,特征833为第三高维特征,将第一高维特征831和第二高维特征832输入至第一注意力机制模型841中,将第一高维特征831和第三高维特征833输入至第二注意力机制模型842中,将第二高维特征832和第三高维特征833输入至第三注意力机制模型843中,以分别得到第一输出特征851、第二输出特征852以及第三输出特征853。
[0164]
将第一输出特征851、第二输出特征852以及第三输出特征853输入至前向神经网络860中,进而得到预测组学特征870,后续对预测组学特征870和第一高维特征831、第二高维特征832以及第三高维特征833分别进行计算,以对标志物发现模型中的参数进行调整,直至训练完毕,在此基础上,后续将多个测试组学特征输入至标志物发现模型后,会得出与多个测试组学特征对应的潜在生物标志物。
[0165]
在本示例性实施例中,将第一高维特征、第二高维特征以及第三高维特征两两组合分别输入至第一注意力机制模型、第二注意力机制模型以及第三注意力机制模型,以捕捉不同组学数据之间的相关性,避免了现有技术未考虑不同组学数据之间相关性的情况发生,提高了生物标志物发现模型的训练准确度,进而提高了后续确定出的潜在生物标志物的准确度。
[0166]
在可选的实施例中,图9示出了生物标志物确定方法中将多个输出特征整合为预测组学特征的流程示意图,如图9所示,该方法至少包括以下步骤:在步骤s910中,将多个输出特征输入至前向神经网络,得到待映射特征。
[0167]
其中,待映射特征为将多个输出特征输入至前向神经网络后,前向神经网络输出的特征。
[0168]
举例而言,如图5所示,多个输出特征包括从注意力机制模型541中输出的特征j1、从注意力机制模型542中输出的特征j2以及从注意力机制模型543中输出的特征j3,将特征
j1、j2以及j3输入前向神经网络后,前向神经网络输出的特征即为待映射特征。
[0169]
在步骤s920中,获取与待映射特征对应的映射公式,并利用映射公式对待映射特征进行计算,得到预测组学特征。
[0170]
其中,映射公式指的是将待映射特征中的元素的值映射至区间(0 1)的公式,进而,利用映射公式对待映射特征进行计算后,得到的预测组学特征中的元素的值的范围在0至1之间。
[0171]
举例而言,映射公式如公式(2)所示。
[0172][0173]
其中,h2为待映射特征,j表示多个输出特征的个数,wj表示与h
2j
计算时使用的权重,b表示一个计算因子,z’为预测组学特征,具体地,如图5所示,由于存在三个输出特征,因此,j为3,h
21
、h
22
以及h
23
分别表示多个输出特征,待映射特征可以为(h
21 h
22 h
23
),。
[0174]
在本示例性实施例中,利用映射公式对待映射特征进行计算可以得到预测组学特征,这为后续确定潜在生物标志物奠定了基础。
[0175]
在可选的实施例中,图10示出了生物标志物确定方法中对生物标志物发现模型进行训练的流程示意图,如图10所示,该方法至少包括以下步骤:在步骤s1010中,获取与待映射特征对应的编码公式,并利用编码公式,将待映射特征转换为预测样本标签。
[0176]
其中,生物标志物发现模型除了可以用于确定出潜在生物标志物之外,还可以用于确定样本属于正常样本还是肿瘤样本,在训练过程中样本指的是训练样本,在预测过程中样本指的是与测试组学数据对应的测试样本。
[0177]
编码公式指的是将待映射特征转化为one-hot编码(一位有效编码向量)的公式,利用编码公式,可以将待映射特征转换为预测样本标签。
[0178]
其中,预测样本标签[1,0]表示预测出与原始组学数据对应的样本为正常样本,预测样本标签[0,1]表示预测出与原始组学数据对应的样本为肿瘤样本。若生物标志物发现模型还用于预测样本的更多分类,例如预测样本为正常样本和具体属于有哪一个肿瘤的样本,则生物标志物发现模型是一个三分类任务,则预测样本标签存在三种形式,分别为[1,0,0]、[0,1,0]以及[0,0,1]。
[0179]
举例而言,编码公式具体可以是利用softmax函数得到的公式,利用编码公式可以将待映射特征转换为预测样本标签。
[0180]
在步骤s1020中,利用预测样本标签,对生物标志物发现模型进行训练。
[0181]
其中,在得到预测样本标签之后,可以利用预测样本标签对生物标志物发现模型进行训练,以使后续利用生物标志物发现模型,预测出与多个测试组学数据对应的预测样本标签。
[0182]
在本示例性实施例中,利用预测样本标签,对生物标志物发现模型进行训练,为后续利用生物标志物发现模型,预测出与多个测试组学数据对应的预测样本标签奠定了基础。
[0183]
在可选的实施例中,图11示出了生物标志物确定方法中对生物标志物发现模型进行训练的流程示意图,如图11所示,该方法至少包括以下步骤:在步骤s1110中,若原始组学数据中包括已知生物标注物,获取与原始组学数据对应的真实样本标签。
[0184]
其中,已知生物标志物指的是已经证明与目标肿瘤相关的生物标志物。由于与原始组学数据对应的样本为已知样本种类的样本,并且,可以通过真实样本标签获知与原始组学数据对应的样本具体属于哪一类样本。
[0185]
举例而言,当原始组学数据中包括已知生物标志物时,此时获取与原始组学数据对应的真实样本标签yi,若训练样本被分为正常样本和肿瘤样本两类,则yi可以为y1或y0。其中,y1表示与该原始组学数据对应的训练样本为肿瘤样本,y0表示与该原始组学数据对应的训练样本为正常样本。
[0186]
在步骤s1120中,确定真实样本标签与预测样本标签之间的第一损失计算关系,并利用第一损失计算关系,对真实样本标签和预测样本标签进行计算得到第一损失计算结果。
[0187]
其中,第一损失计算关系为用于计算真实样本标签和预测样本标签之间的损失值的关系,进而,利用第一损失计算关系,对真实样本标签和预测样本标签进行计算后得到的损失值即为第一损失计算结果。
[0188]
举例而言,第一损失计算关系可以如公式(3)所示。
[0189][0190]
其中,loss1为第一损失计算结果,n为样本类别数,例如当n为2时,由[y0 y1]组成真实样本标签,由[f(x0)f(x1)]组成预测样本标签。
[0191]
在步骤s1130中,确定与已知生物标志物对应的向量与预测组学特征之间的第二损失计算关系,并利用第二损失计算关系,对向量和预测组学特征进行计算得到第二损失计算结果。
[0192]
其中,第二损失计算关系指的是与已知生物标志物对应的向量和预测组学特征之间的损失计算关系。
[0193]
举例而言,确定出的第二损失计算关系可以如公式(4)所示。
[0194]
loss2=-(z*logz’ (1-z)log(1-z’))
ꢀꢀ
(4)
[0195]
其中,loss2为第二损失计算结果,z’为预测组学特征,z为与已知生物标志物对应的向量。将预测组学特征以及与已知生物标志物对应的向量代入公式(4)既可以得到第二损失计算结果。
[0196]
在步骤s1140中,对第一损失计算结果和第二损失结算结果进行计算得到第三损失计算结果,根据第三损失计算结果,对生物标志物发现模型进行训练。
[0197]
其中,第三损失结算结果为对第一损失结算结果和第二损失结算结果进行计算后得到的。
[0198]
基于此,可以利用第三损失计算结果与预设值进行比较,以确定是否还需要对生物标志物发现模型继续进行训练,若第三损失结果小于预设值,则证明生物标志物发现模型的训练精度已经达到预设精度,此时的生物标志物发现模型不需要再继续进行训练,若第三损失计算结果大于或等于预设值,则证明生物标志物发现模型的训练精度还未达到预设精度,此时还需要继续对生物标志物发现模型进行训练。
[0199]
举例而言,可以利用公式(5)对第一损失计算结果和第二损失计算结果进行计算。
[0200]
loss=w1×
loss1 w2×
loss2ꢀꢀ
(5)
[0201]
其中,loss为第三损失计算结构,loss1为第一损失计算结果,loss2为第二损失计
算结果,w1为分配给第一损失计算结果的第一损失权重,w2为分配给第二损失计算结果的第二损失权重。
[0202]
在可选的实施例中,第三损失计算结果是对第一损失计算结果和第二损失计算结果计算后的得到的损失计算结果,进而利用第三损失计算结果对生物标志物发现模型进行训练,有助于确定目标肿瘤与生物标志物之间的第一映射关系,进而保证后续可以基于第一映射关系,在多个测试组学数据中确定出潜在生物标志物。
[0203]
在可选的实施例中,图12示出了生物标志物确定方法中对第一损失计算结果和第二损失计算结果进行计算得到第三损失计算结果的流程示意图,如图12所示,该方法至少包括以下步骤:在步骤s1210中,确定与第一损失结算结果对应的第一损失权重,并确定与第二损失计算结果对应的第二损失权重。
[0204]
其中,第一损失权重与第一损失结果对应,第二损失权重与第二损失计算结果对应,并且,第一损失权重用于调整第一损失计算结果对生物标志物发现模型训练的影响程度,第二损失权重用于调整第二损失计算结果对生物标志物发现模型训练的影响程度。
[0205]
举例而言,与第一损失计算结果loss1对应的第一损失权重为w1,与第二损失计算结果loss2对应的第一损失权重为w2。
[0206]
在步骤s1220中,利用第一损失权重和第二损失权重,对第一损失计算结果和第二损失计算结果进行计算得到第三损失计算结果。
[0207]
其中,第三损失计算结果为利用第一损失权重以及第二损失权重,对第一损失计算结果和第二损失计算结果进行计算后得到的计算结果。
[0208]
举例而言,loss为第三计算结果,则loss=w1×
loss1 w2×
loss2,其中,w1为第一损失权重,w2为第二损失权重,loss1为第一损失计算结果,loss2为第二损失计算结果。
[0209]
在可选的示例性实施例中,利用第一损失权重、第二损失权重,对第一损失计算结果和第二损失计算结果进行计算得到第三损计算结果,有助于根据不同的第一损失权重以及不同的第二损失权重,确定出不同的第三损失计算结果,进而可以灵活地调整第一损失权重以及第二损失权重,以对生物标志物发现模型进行灵活的训练。
[0210]
在步骤s130中,基于第一映射关系在多个测试组学数据中确定出潜在生物标志物。
[0211]
在本公开的示例性实施例中,测试组学数据指的是需要输入至生物标志物发现模型中,以发现测试组学数据中可能存在的潜在生物标志物的数据。
[0212]
举例而言,测试组学数据中包括基因突变测试组学数据、基因甲基测试组学数据以及蛋白测试组学数据,将上述测试组学数据输入至已经训练完成的生物标志物发现模型中后,生物标志物发现模型会根据之前确定出的第一映射关系,在测试组学数据中确定出潜在生物标志物,具体地,潜在生物标志物可以是基因k。
[0213]
在可选的实施例中,图13示出了生物标志物确定方法中确定出潜在生物标志物的流程示意图,如图13所示,该方法至少包括以下步骤:在步骤s1310中,基于第一映射关系,确定出与测试组学数据对应的预测组学特征。
[0214]
其中,预测组学特征指的是将测试组学数据输入至训练完成的生物标志物发现模型后的得到的特征。
[0215]
举例而言,将预测组学数据输入至训练完成的生物标志物发现模型,可以得到预
测组学特征。
[0216]
在步骤s1320中,确定与预测组学特征对应的特征元素,对特征元素的大小进行比较得到元素比较结果。
[0217]
其中,预测组学特征是由特征元素组成的,元素比较结果即对特征元素的大小进行比较后得到的结果。
[0218]
举例而言,预测组学特征为[0.03 0.05 0.09],则与预测组学特征对应的特征元素为0.03、0.05以及0.09,进而得到的元素比较结果为0.09大于0.05,0.05大于0.03。
[0219]
在步骤s1330中,根据元素比较结果,确定出最大特征元素,以将与最大特征元素对应的生物标志物确定为潜在生物标志物;其中,潜在生物标志物包括基因、甲基化基因、蛋白质中的任意一项。
[0220]
其中,潜在生物标志物为与最大特征元素对应的生物标志物,具体地,若与最大特征元素对应的生物标志物为某一基因,则该基因即为潜在生物标志物;若与最大特征元素对应的生物标志物为某一甲基化基因,则该甲基化基因即为潜在生物标志物;若与最大特征元素对应的生物标志物为某一蛋白质,则该蛋白质即为潜在生物标志物。
[0221]
举例而言,元素比较结果为0.09大于0.05,0.05大于0.03,则最大特征元素为0.09,并且,与最大特征元素对应的生物标志物为基因a,则基因a即为潜在生物标志物。
[0222]
在本示例性实施例中,基于第一映射关系,可以得到与测试组学数据对应的预测组学特征,进而根据预测组学特征可以确定出潜在生物标志物,避免了现有技术中将突变频率高的生物标志物确定为潜在生物标志物的情况发生,提高了确定出的潜在生物标志物准确度。
[0223]
本公开还提出了一种肿瘤样本筛查方法。图14示出了肿瘤样本筛查方法的流程示意图,如图14所示,肿瘤样本筛查方法至少包括以下步骤:
[0224]
步骤s1410.获取与目标肿瘤对应的训练样本,并将与训练样本对应的原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,确定出训练样本与样本标签之间的第二映射关系。
[0225]
步骤s1420.获取与目标肿瘤对应的测试样本,并将与测试样本对应的测试组学数据输入至生物标志物发现模型中。
[0226]
步骤s1430.基于第二映射关系,确定出与测试样本对应的样本标签,以根据样本标签在测试样本中筛查出肿瘤样本。
[0227]
在本公开的示例性实施例提供的方法及装置中,根据与目标肿瘤具有对应关系的生物标志物发现模型筛查出肿瘤样本,一方面,生物标志物发现模型是将原始组学数据作为训练数据进行训练得到的,并且,原始组学数据与目标肿瘤对应,进而,可以筛查出与不同目标肿瘤对应的肿瘤样本,适用于所有不同的目标肿瘤;另一方面,避免了现有技术中,将突变频率高的基因作为筛查肿瘤样本的依据,提高了确定出的肿瘤样本的准确度。
[0228]
下面对肿瘤样本筛查方法的各个步骤进行详细说明。
[0229]
在步骤s1410中,获取与目标肿瘤对应的训练样本,并将与训练样本对应的原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,确定出训练样本与样本标签之间的第二映射关系。
[0230]
其中,目标肿瘤与步骤s110中的相同,生物标志物发现模型如图5所示,训练样本
指的是与目标肿瘤对应的,输入生物标志物发现模型之中,以对生物标志物模型进行训练的样本。值得说明的是,训练样本中包括正常样本以及肿瘤样本。原始组学特征与步骤s110中的原始组学特征一致,样本标签指的是表示训练样本属于正常样本还是肿瘤样本的标签,并且,若训练样本属于肿瘤样本,则样本标签还可以用来表示肿瘤样本具体属于哪一种肿瘤。
[0231]
第二映射关系,指的是训练样本与样本标签之间的对应关系。
[0232]
举例而言,目标肿瘤为子宫癌,将与子宫癌对应的原始组学数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,在训练完成后,确定出了训练样本与样本标签之间第二的映射关系。其中,样本标签用于表示训练样本属于正常样本还是与患有子宫癌的患者对应的肿瘤样本。
[0233]
在可选的实施例中,图15示出了肿瘤样本筛查方法中对生物标志物发现模型进行训练的流程示意图,如图15所示,该方法至少包括以下步骤:在步骤s1510中,若原始组学数据中不包括已知生物标注物,获取与原始组学数据对应的真实样本标签。
[0234]
其中,已知生物标志物指的是已经证明与目标肿瘤相关的生物标志物。由于与多个原始组学数据对应的样本为已知样本种类的样本,并且,可以通过真实样本标签获知与多个原始组学数据对应的样本具体属于哪一类样本。
[0235]
举例而言,当多个原始组学数据中不包括已知生物标志物时,此时获取与多个原始组学数据对应的真实样本标签yi。
[0236]
在步骤s1520中,确定真实样本标签与预测样本标签之间的第一损失计算关系,并利用第一损失计算关系,对真实样本标签和预测样本标签进行计算得到第一损失计算结果。
[0237]
其中,预测样本标签为将测试样本的测试组学数据数据输入至生物标志物发现模型当中得到的,生物标志物发现模型的模型结构如图5所示,在得到待映射特征之后,可以利用编码公式计算出预测样本标签,编码公式指的是将待映射特征转化为one-hot编码(一位有效编码向量)的公式。
[0238]
举例而言,第一损失计算关系为用于计算真实样本标签和预测样本标签之间的损失值的关系,进而,利用第一损失计算关系,对真实样本标签和预测样本标签进行计算后得到的损失值即为第一损失计算结果。
[0239]
举例而言,第一损失计算关系可以如公式(3)所示。
[0240]
在步骤s1530中,根据第一损失计算结果,对生物标志物发现模型进行训练,以确定出训练样本与样本标签之间的第二映射关系。
[0241]
其中,在计算出第一损失计算结果之后,对生物标志物发现模型进行训练,直至训练结束,进而确定出训练样本与样本标签之间的第二映射关系。
[0242]
通常而言,训练结束的条件为训练次数达到预设次数,或对预测样本标签与真实样本标签进行损失计算得到的第一损失计算结果小于预设值。
[0243]
举例而言,预测样本标签为[1,0],真实样本标签为[1,0],利用第一损失计算关系,对预测样本标签和真实样本标签进行损失计算得到第一损失计算结果,以利用第一损失计算结果对生物标志物发现模型进行训练。
[0244]
在可选的实施例中,根据第一损失计算结果对生物标志物发现模型进行训练,进
而在原始组学数据中不包括生物标志物时,生物标志物发现模型可以用于预测与测试样本对应的样本标签,扩大了生物标志物发现模型的使用场景。
[0245]
在步骤s1420中,获取与目标肿瘤对应的测试样本,并将与测试样本对应的测试组学数据输入至生物标志物发现模型中。
[0246]
其中,测试样本与步骤s120中的测试样本相同,测试组学数据与步骤s120中的测试组学数据相同。
[0247]
举例而言,获取到与子宫癌对应的测试组学数据,并将该测试组学数据输入至生物标志物发现模型中。
[0248]
在步骤s1430中,基于第二映射关系,确定出与训练样本对应的样本标签,以根据样本标签在测试样本中筛查出肿瘤样本。
[0249]
其中,测试组学数据需要输入至生物标志物发现模型中,以从与测试组学数据对应测试样本中筛查出肿瘤样本。
[0250]
举例而言,测试组学数据中包括基因突变测试组学数据、基因甲基测试组学数据以及蛋白测试组学数据,将上述多个测试组学数据输入至已经训练完成的生物标志物发现模型中后,生物标志物发现模型会根据之前确定出的第二映射关系,在测试样本中筛查出肿瘤样本。
[0251]
在本公开的示例性实施例提供的方法及装置中,根据与目标肿瘤具有对应关系的生物标志物发现模型筛查出肿瘤样本,一方面,生物标志物发现模型是将原始组学数据作为训练数据进行训练得到的,并且,原始组学数据与目标肿瘤对应,进而,可以筛查出与不同目标肿瘤对应的肿瘤样本,适用于所有不同的目标肿瘤;另一方面,避免了现有技术中,将突变频率高的突变基因作为筛查肿瘤样本的依据,提高了确定出的肿瘤样本的准确度。
[0252]
下面结合一应用场景对本公开实施例中生物标志物确定方法做出详细说明。
[0253]
目标肿瘤为具有相关性的乳腺癌和卵巢癌,基于此,获取与乳腺癌对应的原始组学数据aa以及与卵巢癌对应的原始组学数据bb,并将原始组学数据aa以及原始组学数据bb输入至生物标志物发现模型xx中,以对生物标志物发现模型进行训练,进而确定与生物标志物与上述两种目标肿瘤之间的第一映射关系。
[0254]
获取与乳腺癌对应的测试组学数据cc,并将测试组学数据cc输入至生物标志物发现模型xx中,以基于第一映射关系确定出在测试组学数据cc中存在的潜在生物标志物。
[0255]
在本应用场景中,根据与目标肿瘤具有对应关系的生物标志物发现模型确定出潜在生物标志物,一方面,生物标志物发现模型是将原始组学数据作为训练数据进行训练得到的,并且,原始组学数据与目标肿瘤对应,进而,可以确定出与不同目标肿瘤对应的不同的潜在生物标志物,适用于所有不同的目标肿瘤;另一方面,避免了现有技术中,将突变频率高的突变基因作为生物标志物的情况发生,提高了确定出的潜在生物标志物的准确度。
[0256]
此外,在本公开的示例性实施例中,还提供一种生物标志物确定装置。图16示出了生物标志物确定装置的结构示意图,如图16所示,生物标志物确定装置1600可以包括:第一训练模块1610、第一输入模块1620和预测模块1630。其中:
[0257]
第一训练模块1610,被配置为获取与目标肿瘤对应的原始组学数据,并将原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,确定出目标肿瘤和生物标志物之间的第一映射关系;第一输入模块1620,被配置为获取与
目标肿瘤对应的测试组学数据,并将测试组学数据输入至生物标志物发现模型中;预测模块1630,被配置为基于第一映射关系在测试组学数据中确定出潜在生物标志物。
[0258]
上述生物标志物确定装置1600的具体细节已经在对应的生物标志物确定方法中进行了详细的描述,因此此处不再赘述。
[0259]
在本公开的示例性实施例中,还提供一种肿瘤样本筛查装置。图17示出了肿瘤样本筛查装置的结构示意图,如图17所示,肿瘤样本筛查装置1700可以包括:第二训练模块1710、第二输入模块1720和筛查模块1730。其中:
[0260]
第二训练模块1710,被配置为获取与目标肿瘤对应的训练样本,并将与训练样本对应的原始组学数据作为训练数据输入至生物标志物发现模型中,以对生物标志物发现模型进行训练,确定出目标肿瘤与样本标签之间的第二映射关系;第二输入模块1720,被配置为获取与目标肿瘤对应的测试样本,并将与测试样本对应的测试组学数据输入至生物标志物发现模型中;筛查模块1730,被配置为基于第二映射关系,确定出与训练样本对应的样本标签,以根据样本标签在测试样本中筛查出肿瘤样本。
[0261]
应当注意,尽管在上文详细描述中提及生物标志物确定装置1600以及肿瘤样本筛查装置1700的若干模块或者单元,但是这种划分并非强制性的。实际上,根据本公开的实施方式,上文描述的两个或更多模块或者单元的特征和功能可以在一个模块或者单元中具体化。反之,上文描述的一个模块或者单元的特征和功能可以进一步划分为由多个模块或者单元来具体化。
[0262]
此外,在本公开的示例性实施例中,还提供了一种能够实现上述方法的电子设备。
[0263]
下面参照图18来描述根据本发明的这种实施例的电子设备1800。图18显示的电子设备1800仅仅是一个示例,不应对本发明实施例的功能和使用范围带来任何限制。
[0264]
如图18所示,电子设备1800以通用计算设备的形式表现。电子设备1800的组件可以包括但不限于:上述至少一个处理单元1810、上述至少一个存储单元1820、连接不同系统组件(包括存储单元1820和处理单元1810)的总线1830、显示单元1840。
[0265]
其中,所述存储单元存储有程序代码,所述程序代码可以被所述处理单元1810执行,使得所述处理单元1810执行本说明书上述“示例性方法”部分中描述的根据本发明各种示例性实施例的步骤。
[0266]
存储单元1820可以包括易失性存储单元形式的可读介质,例如随机存取存储单元(ram)1821和/或高速缓存存储单元1822,还可以进一步包括只读存储单元(rom)1823。
[0267]
存储单元1820还可以包括具有一组(至少一个)程序模块1825的程序/使用工具1824,这样的程序模块1825包括但不限于:操作系统、一个或者多个应用程序、其它程序模块以及程序数据,这些示例中的每一个或某种组合中可能包含网络环境的现实。
[0268]
总线1830可以为表示几类总线结构中的一种或多种,包括存储单元总线或者存储单元控制器、外围总线、图形加速端口、处理单元或者使用多种总线结构中的任意总线结构的局域总线。
[0269]
电子设备1800也可以与一个或多个外部设备1870(例如键盘、指向设备、蓝牙设备等)通信,还可与一个或者多个使得用户能与该电子设备1800交互的设备通信,和/或与使得该电子设备1800能与一个或多个其它计算设备进行通信的任何设备(例如路由器、调制解调器等等)通信。这种通信可以通过输入/输出(i/o)接口1850进行。并且,电子设备1800
还可以通过网络适配器1860与一个或者多个网络(例如局域网(lan),广域网(wan)和/或公共网络,例如因特网)通信。如图所示,网络适配器1860通过总线1830与电子设备1800的其它模块通信。应当明白,尽管图中未示出,可以结合电子设备1800使用其它硬件和/或软件模块,包括但不限于:微代码、设备驱动器、冗余处理单元、外部磁盘驱动阵列、raid系统、磁带驱动器以及数据备份存储系统等。
[0270]
通过以上的实施例的描述,本领域的技术人员易于理解,这里描述的示例实施例可以通过软件实现,也可以通过软件结合必要的硬件的方式来实现。因此,根据本公开实施例的技术方案可以以软件产品的形式体现出来,该软件产品可以存储在一个非易失性存储介质(可以是cd-rom,u盘,移动硬盘等)中或网络上,包括若干指令以使得一台计算设备(可以是个人计算机、服务器、终端装置、或者网络设备等)执行根据本公开实施例的方法。
[0271]
在本公开的示例性实施例中,还提供了一种计算机可读存储介质,其上存储有能够实现本说明书上述方法的程序产品。在一些可能的实施例中,本发明的各个方面还可以实现为一种程序产品的形式,其包括程序代码,当所述程序产品在终端设备上运行时,所述程序代码用于使所述终端设备执行本说明书上述“示例性方法”部分中描述的根据本发明各种示例性实施例的步骤。
[0272]
参考图19所示,描述了根据本发明的实施例的用于实现上述方法的程序产品1900,其可以采用便携式紧凑盘只读存储器(cd-rom)并包括程序代码,并可以在终端设备,例如个人电脑上运行。然而,本发明的程序产品不限于此,在本文件中,可读存储介质可以是任何包含或存储程序的有形介质,该程序可以被指令执行系统、装置或者器件使用或者与其结合使用。
[0273]
所述程序产品可以采用一个或多个可读介质的任意组合。可读介质可以是可读信号介质或者可读存储介质。可读存储介质例如可以为但不限于电、磁、光、电磁、红外线、或半导体的系统、装置或器件,或者任意以上的组合。可读存储介质的更具体的例子(非穷举的列表)包括:具有一个或多个导线的电连接、便携式盘、硬盘、随机存取存储器(ram)、只读存储器(rom)、可擦式可编程只读存储器(eprom或闪存)、光纤、便携式紧凑盘只读存储器(cd-rom)、光存储器件、磁存储器件、或者上述的任意合适的组合。
[0274]
计算机可读信号介质可以包括在基带中或者作为载波一部分传播的数据信号,其中承载了可读程序代码。这种传播的数据信号可以采用多种形式,包括但不限于电磁信号、光信号或上述的任意合适的组合。可读信号介质还可以是可读存储介质以外的任何可读介质,该可读介质可以发送、传播或者传输用于由指令执行系统、装置或者器件使用或者与其结合使用的程序。
[0275]
可读介质上包含的程序代码可以用任何适当的介质传输,包括但不限于无线、有线、光缆、rf等等,或者上述的任意合适的组合。
[0276]
可以以一种或多种程序设计语言的任意组合来编写用于执行本发明操作的程序代码,所述程序设计语言包括面向对象的程序设计语言—诸如java、c 等,还包括常规的过程式程序设计语言—诸如“c”语言或类似的程序设计语言。程序代码可以完全地在用户计算设备上执行、部分地在用户设备上执行、作为一个独立的软件包执行、部分在用户计算设备上部分在远程计算设备上执行、或者完全在远程计算设备或服务器上执行。在涉及远程计算设备的情形中,远程计算设备可以通过任意种类的网络,包括局域网(lan)或广域网
(wan),连接到用户计算设备,或者,可以连接到外部计算设备(例如利用因特网服务提供商来通过因特网连接)。
[0277]
本领域技术人员在考虑说明书及实践这里公开的发明后,将容易想到本公开的其他实施例。本技术旨在涵盖本公开的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本公开的一般性原理并包括本公开未公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为示例性的,本公开的真正范围和精神由权利要求指出。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
