1.本发明涉及医药,特别是一种从鄂西香茶菜中提取的化合物1和化合物2及其制备方法与应用。
背景技术:
2.鄂西香茶菜为唇形科香茶菜属植物,分布于我国的河南省西部、湖北省的西部及河北省南部等地,被当地人用作抗菌、抗炎和抗癌药物使用。其性凉,味辛、苦;归肝、肾经;有清热利湿、解毒消肿等功效。近年来,唇形科香茶菜属植物因含有结构新颖和生物活性好的二萜类成分而备受关注,部分化合物作为抗肿瘤候选化合物已进入研发阶段,如冬凌草甲素,毛萼香茶菜甲素等。遵循传统用法,同时结合对鄂西香茶菜的化学成分及活性研究,希望获得更多结构新颖、抗炎活性显著的新化合物。现已有鄂西香茶菜素作为wnt信号通路抑制剂及抗癌药物的应用(申请号201310185250.2)、sars等冠状病毒主蛋白酶的二萜类天然产物抑制剂及其筛选方法(申请号200710195754.7)中记载从鄂西香茶菜中筛选二萜类天然产物抑制剂的方法,但至今未见有从鄂西香茶菜制备具有抗炎活性的化合物1(isohenolide a)和化合物2(isohenolide b),也未有鄂西香茶菜中哪些活性成分具有抗炎活性,并且如何从鄂西香茶菜中提出抗炎活性成分的公开报导。
技术实现要素:
3.针对上述情况,为克服现有技术之缺陷,本发明之目的就是提供一种从鄂西香茶菜中提取的化合物1和化合物2及其制备方法与应用,可有效解决从鄂西香茶菜中制备化合物1和化合物2,实现在制备抗炎药物中的应用问题。
4.本发明解决的技术方案是,一种从鄂西香茶菜中提取的化合物1和化合物2,其分子式分别为c
22h28
o7和c
20h28
o6,不饱和度分别为9和7,结构式分别为:
[0005][0006]
其制备方法包括以下步骤:
[0007]
(1)取干燥的鄂西香茶菜全草15kg,每次加体积浓度50~95%乙醇100~500l,回流提取1~4次,每次提取1~4h,合并提取液,减压浓缩至相当于生药量0.5~2.0g/ml的样品溶液;
[0008]
(2)在步骤(1)的样品溶液中加水5~10l分散成悬浮液,先用石油醚重复萃取3~10次,每次用量10~30l,充分振摇5min,静置分层,弃去石油醚层;水层继续用乙酸乙酯萃取3~8次,每次用量5~30l,充分振摇5min,静置分层,合并乙酸乙酯层,减压回收溶剂,得到干燥固体组分fr.
etoac
(81.1~116.6g);
[0009]
(3)组分fr.
etoac
经80~300目硅胶柱色谱,硅胶柱r=2~8cm、h=10~50cm,用体积比100︰0、100︰2、100︰5、0︰1的石油醚-丙酮进行梯度洗脱,每个比例洗脱2~10个柱体积,收集100︰5的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6(4.1~8.0g);
[0010]
(4)将组分fr.
etoac-6用体积比1︰1的二氯甲烷-甲醇溶解,经sephadex lh-20柱层析,柱r=1.0~2.0cm、h=60~120cm,洗脱溶剂为体积比1︰1的二氯甲烷-甲醇,流速0.2~0.6ml/min,tlc检识合并第950~2450min的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6-b(1.6~3.2g);
[0011]
(5)将组分fr.
etoac-6-b用甲醇溶解成50mg/ml的样品溶液,用ods-aq柱在半制备hplc上进一步分离,洗脱溶剂为体积比25︰75的乙腈-水,流速3ml/min,分别收集30.23min、45.16min的色谱峰,减压回收溶剂,得到干燥粉末状的化合物1(47.0~80.2mg)和化合物2(39.6~59.4mg)。
[0012]
本发明原料丰富,制备方法易操作,制备的产物经鉴定,是从鄂西香茶菜提取的具有抗炎活性的新化合物,该化合物可有效用于制备抗炎的药物,开拓了鄂西香茶菜的药用价值和商业价值,经济和社会效益巨大。
附图说明
[0013]
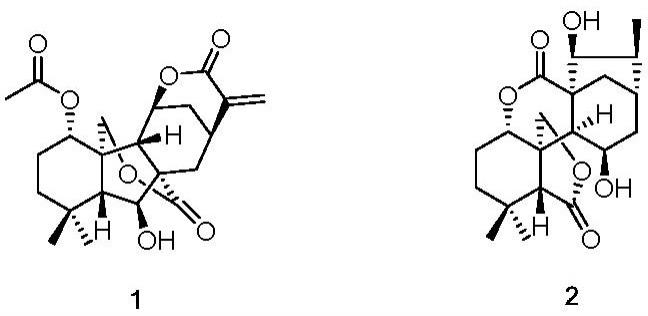
图1为本发明化合物1、化合物2的分子结构式图。
[0014]
图2为本发明化合物1、化合物2的关键hmbc相关图。
[0015]
图3为本发明化合物1、化合物2的关键roesy相关图。
[0016]
图4为本发明化合物1、化合物2的ecd相关图。
[0017]
图5为本发明的工艺流程图。
具体实施方式
[0018]
以下结合具体情况对本发明的具体实施方式作详细说明。
[0019]
本发明在具体实施中可由以下实施例给出。
[0020]
实施例1
[0021]
本发明一种从鄂西香茶菜中提取的化合物1和化合物2的制备方法,包括以下步骤:
[0022]
(1)取干燥的鄂西香茶菜全草15kg,每次加体积浓度70%乙醇300l,回流提取2次,每次提取2h,合并提取液,减压浓缩至相当于生药量1.5g/ml的样品溶液;
[0023]
(2)在步骤(1)的样品溶液中加水5l分散成悬浮液,先用石油醚重复萃取8次,每次用量20l,充分振摇5min,静置分层,弃去石油醚层;水层继续用乙酸乙酯重复萃取6次,每次用量20l,充分振摇5min,静置分层,合并乙酸乙酯层,减压回收溶剂,得到干燥固体组分fr.
etoac
(101.5g);
[0024]
(3)组分fr.
etoac
经200目硅胶柱色谱,硅胶柱r=4cm、h=20cm,用体积比100︰0、
100︰2、100︰5、0︰1的石油醚-丙酮进行梯度洗脱,每个比例洗脱5个柱体积,收集100︰5的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6(6.3g);
[0025]
(4)将组分fr.
etoac-6用体积比1︰1的二氯甲烷-甲醇溶解,经sephadex lh-20柱层析,柱r=1.3cm、h=90cm,洗脱溶剂为体积比1︰1的二氯甲烷-甲醇,流速0.3ml/min,tlc检识合并第1350~1800min的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6-b(2.5g);
[0026]
(5)将组分fr.
etoac-6-b用甲醇溶解成50mg/ml的样品溶液,用ods-aq柱在半制备hplc上进一步分离,洗脱溶剂为体积比25︰75的乙腈-水,流速3ml/min,分别收集30.23min、45.16min的色谱峰,减压回收溶剂,得到干燥粉末状的化合物1(73.0mg)和化合物2(55.6mg)。
[0027]
实施例2
[0028]
本发明一种从鄂西香茶菜中提取的化合物1和化合物2的制备方法,包括以下步骤:
[0029]
(1)取干燥的鄂西香茶菜全草15kg,每次加体积浓度95%乙醇100l,回流提取4次,每次提取1h,合并提取液,减压浓缩至相当于生药量2.0g/ml的样品溶液;
[0030]
(2)在步骤(1)的样品溶液中加水10l分散成悬浮液,先用石油醚重复萃取10次,每次用量25l,充分振摇5min,静置分层,弃去石油醚层;水层继续用乙酸乙酯重复萃取8次,每次用量25l,充分振摇5min,静置分层,合并乙酸乙酯层,减压回收溶剂,得到干燥固体组分fr.
etoac
(116.6g);
[0031]
(3)组分fr.
etoac
经80目硅胶柱色谱,硅胶柱r=6cm、h=10cm,用体积比100︰0、100︰2、100︰5、0︰1的石油醚-丙酮进行梯度洗脱,每个比例洗脱10个柱体积,收集100︰5的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6(8.0g);
[0032]
(4)将组分fr.
etoac-6用体积比1︰1的二氯甲烷-甲醇溶解,经sephadex lh-20柱层析,柱r=2.0cm、h=60cm,洗脱溶剂为体积比1︰1的二氯甲烷-甲醇,流速0.6ml/min,tlc检识合并第950~1130min的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6-b(3.2g);
[0033]
(5)将组分fr.
etoac-6-b用甲醇溶解成50mg/ml的样品溶液,用ods-aq柱在半制备hplc上进一步分离,洗脱溶剂为体积比25︰75的乙腈-水,流速3ml/min,分别收集30.23min、45.16min的色谱峰,减压回收溶剂,得到干燥粉末状的化合物1(80.2mg)和化合物2(59.4mg)。
[0034]
实施例3
[0035]
本发明一种从鄂西香茶菜中提取的化合物1和化合物2的制备方法,包括以下步骤:
[0036]
(1)取干燥的鄂西香茶菜全草15kg,每次加体积浓度50%乙醇500l,回流提取1次,提取4h,合并提取液,减压浓缩至相当于生药量0.6g/ml的样品溶液;
[0037]
(2)在步骤(1)的样品溶液中加水5l分散成悬浮液,先用石油醚重复萃取3次,每次用量30l,充分振摇5min,静置分层,弃去石油醚层;水层继续用乙酸乙酯重复萃取3次,每次用量30l,充分振摇5min,静置分层,合并乙酸乙酯层,合并乙酸乙酯萃取溶液,减压回收溶剂,得到干燥固体组分fr.
etoac
(81.1g);
[0038]
(3)组分fr.
etoac
经300目硅胶柱色谱,硅胶柱r=2cm、h=50cm,用体积比100︰0、100︰2、100︰5、0︰1的石油醚-丙酮进行梯度洗脱,每个比例洗脱2个柱体积,收集100︰5的洗
脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6(4.1g);
[0039]
(4)将组分fr.
etoac-6用体积比1︰1的二氯甲烷-甲醇溶解,经sephadex lh-20柱层析,柱r=1.0cm、h=120cm,洗脱溶剂为体积比1︰1的二氯甲烷-甲醇,流速0.2ml/min,tlc检识合并第1890~2450min的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6-b(1.6g);
[0040]
(5)将组分fr.
etoac-6-b用甲醇溶解成50mg/ml的样品溶液,用ods-aq柱在半制备hplc上进一步分离,洗脱溶剂为体积比25︰75的乙腈-水,流速3ml/min,分别收集30.23min、45.16min的色谱峰,减压回收溶剂,得到干燥粉末状的化合物1(47.0mg)和化合物2(39.6mg)。
[0041]
实施例4
[0042]
本发明一种从鄂西香茶菜中提取的化合物1和化合物2的制备方法,包括以下步骤:
[0043]
(1)取干燥的鄂西香茶菜全草15kg,每次加体积浓度80%乙醇300l,回流提取3次,每次提取2h,合并提取液,减压浓缩至相当于生药量1.8g/ml的样品溶液;
[0044]
(2)在步骤(1)的样品溶液中加水8l分散成悬浮液,先用石油醚重复萃取8次,每次用量22l,充分振摇5min,静置分层,弃去石油醚层;水层继续用乙酸乙酯重复萃取6次,每次用量22l,充分振摇5min,静置分层,合并乙酸乙酯层,减压回收溶剂,得到干燥固体组分fr.
etoac
(109.7g);
[0045]
(3)组分fr.
etoac
经160目硅胶柱色谱,硅胶柱r=5cm、h=20cm,用体积比100︰0、100︰2、100︰5、0︰1的石油醚-丙酮进行梯度洗脱,每个比例洗脱6个柱体积,收集100︰5的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6(7.1g);
[0046]
(4)将组分fr.
etoac-6用体积比1︰1的二氯甲烷-甲醇溶解,经sephadex lh-20柱层析,柱r=1.5cm、h=80cm,洗脱溶剂为体积比1︰1的二氯甲烷-甲醇,流速0.4ml/min,tlc检识合并第1300~1570min的洗脱液,减压回收溶剂,得到干燥粉末组分fr.
etoac-6-b(2.7g);
[0047]
(5)将组分fr.
etoac-6-b用甲醇溶解成50mg/ml的样品溶液,用ods-aq柱在半制备hplc上进一步分离,洗脱溶剂为体积比25︰75的乙腈-水,流速3ml/min,分别收集30.23min、45.16min的色谱峰,减压回收溶剂,得到干燥粉末状的化合物1(71.5mg)和化合物2(56.0mg)。
[0048]
要指出的是,上述给出的实施例仅是用于说明本发明的具体实施方式,以对该从鄂西香茶菜中提取具有抗炎活性的化合物及其提取方法进行的详细描述,是说明性的,而不是用于限定本发明的保护范围,凡是在不脱离本发明总体构思下的变化和修改、等同等效替换,均应属本发明的保护范围之内。
[0049]
上述所得化合物经测定,鉴定为从鄂西香茶菜中提取的两个新化合物:化合物1(isohenolide a)、化合物2(isohenolide b),分子结构式见图1所示。对化合物1、化合物2进行活性实验研究,表明其具有抗炎活性。有关具体测定和实验资料如下:
[0050]
一、结构鉴定
[0051]
经结构鉴定,实施例1-4给出的化合物1和化合物2为从鄂西香茶菜中提取的两个活性成分,均是结构分别相同的两个新化合物,具体鉴定情况如下:
[0052]
1、化合物1:白色无定型粉末,纯度为96.7%,易溶于甲醇等有机溶剂。旋光度[α]
2d0
10(c 0.01,meoh);紫外光谱显示在204nm有最大吸收峰;红外光谱显示有羟基(3338cm-1
),
15.333(c 0.01,meoh);紫外光谱显示在202nm有最大吸收峰;红外光谱显示有羟基(3389cm-1
),羰基(1651cm-1
)的存在;在hresims中准分子离子峰[m cooh]
—
为m/z 409.1865(c
21h29
o8计算值为409.1870);结合1h-nmr、
13
c-nmr谱确定分子式为c
20h28
o6,不饱和度为7。
[0059]1h-nmr和hsqc谱显示有一个连氧亚甲基δ
h 4.19(d,j=9.2hz,h-20),3.70(d,j=9.2hz,h-20);三个连氧次甲基δ
h 4.68(m,h-15),4.66(m,h-1),3.77(dp,j=13.5,7.7,6.7hz,h-11);四个次甲基δ
h 3.50(m,h-5),2.24(d,j=9.8hz,h-9),2.11(m,h-13),2.16(m,h-16);三个甲基δ
h 1.07(s,h
3-18),0.93(s,h
3-19),0.80(d,j=7.1hz,h
3-17)。
13
c-nmr和hsqc谱显示有20个碳信号,其中有两个酯羰基δ
c 176.8(c-6),174.9(c-7);一个连氧亚甲基δ
c 70.2(c-20);三个连氧次甲基δ
c 74.2(c-1),73.0(c-15),61.6(c-11);三个次甲基δ
c 49.9(c-5),44.0(c-9),37.3(c-16),36.4(c-13);三个甲基δ
c 32.2(c-18),23.8(c-19),11.8(c-22)。详细信息见表1。以上数据提示该化合物可能是一个贝壳杉烷型二萜类化合物。
[0060]1h-1
h cosy谱显示显示出两个自旋耦合片段h-1/h
2-2/h
2-3和h-9/h-11/h
2-12/h-13/h
2-14/h-15/h-16/h
3-17(见图2)。hmbc谱显示关键的h
2-20与co-6相关,由h-1与co-7相关,提示c-6位与c-20位,c-1位与c-7位之间分别存在一个内酯键连接。由甲基h
3-17与c-13、c-15、c-16相关;连氧次甲基质子h-15与c-7、c-8、c-9、c-14相关,h-11与c-8、c-9、c-10、c-12、c-13相关,确定c-17位为甲基,且c-11位和c-15位分别有游离羟基取代。结合1d和2d nmr确定了化合物2的平面结构,为延命草素型贝壳杉烷类二萜。
[0061]
化合物2的相对构型是通过roesy分析确定的。由h-5与h-1、h
3-18相关,h
2-20与h-9、h
3-19相关,提示h-1、h-5和me-18处于同一侧,暂定为β构型;而h-9、h
3-19以及ch
2-20应处于另一侧,即α构型。由h-1与h-14相关,h-11与h-9、h-16相关,h-15与h-16相关,h-13与h
3-17相关,提示h-11、h-15、h-16为α构型;h-13与ch
2-14片段为β构型。
[0062]
化合物2的绝对构型是通过对比实测和计算ecd谱确定的。ecd光谱在200~250nm处表现出负的科顿效应。计算曲线与实验曲线吻合良好,但有轻微的峰移(见图4),表明化合物2的绝对构型为1s,5r,8s,9s,10s,11r,13s,15s,16s,分子结构式为:
[0063][0064]
表1.化合物1和化合物2的1h和
13
c-nmr数据(500and 125mhz,δin ppm,j in hz)
[0065][0066][0067]acompound 1was recorded in cdcl3,bcompound 2was recorded in dmso-d6.
[0068]
二、活性实验
[0069]
以化合物1和化合物2为主成分制备了一种治疗急性咽炎的药物,进行了体内抗炎实验。
[0070]
试验方法:30只sd大鼠,雌雄各半,体质量(200
±
20)g,动物由济南朋悦实验动物繁育有限公司提供,实验动物许可证号:syxk(鲁)20190003。适应性喂养1周,随机分为5组,每组6只。模型组和各给药组以喉头喷雾器将15%的氨水向大鼠咽部喷雾散布(面积150mm2),每次喷3下(0.2~0.4ml),每天两次,连续3d,使大鼠咽部黏膜因氨水急性刺激而充血肿胀,形成急性炎症。对照组大鼠给予等量蒸馏水。造模成功后第2d给药。给药组分别灌服高、低剂量(150mg/kg、75mg/kg)制备的药物,阳性对照组(慢严舒柠清喉利咽颗粒,3.0g/kg),对照组与模型组灌服等量生理盐水,连续给药7d。实验过程中每天观察和记录各组动物饮食、饮水变化、活动状态及大鼠咽部变化情况,包括黏膜形态和色泽、口腔分泌物。
[0071]
制备的抗炎药物高、低剂量组均有显著的抗炎作用。模型组大鼠在造模后第2天出现挠抓口部,观察大鼠咽部,出现充血、肿胀、分泌物增多,饮水频率和水量增加,食量下降
等现象。造模第3天,除上述症状更加明显外,大鼠唇周出现抓挠血痕,口腔内有溃烂现象,大鼠精神萎靡,体温稍低,活动量减少。对照组大鼠造模2-4天期间均未出现上述现象。给药2d后,高剂量组和各给药组大鼠纳食量开始增大,咽部分泌物减少,口腔内溃烂现象明显好转,精神基本恢复正常,且高剂量组疗效优于低剂量组,5d后基本痊愈;而模型组大鼠在5d左右症状有轻微改善。提示制备的抗炎药物在150mg/kg和75mg/kg剂量下均可以显著抑制氨水喷雾法致大鼠急性咽炎,具有很好的抗炎活性。
[0072]
因此,本发明化合物1、化合物2作为抗炎先导化合物,有效用于制备抗炎药物,实现在制备治疗咽炎药物中的应用,开拓了鄂西香茶菜的药用价值和商业价值,有巨大的开发应用前景,经济和社会效益巨大。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。