1.本发明涉及功能材料技术领域,具体涉及一种有机硅季铵盐双胍盐材料、制备方法和抗菌应用。
背景技术:
2.在人们生活的环境中存在着有害细菌、真菌和病毒等种类繁多的微生物,而这些有害的微生物正是导致人们患上疾病的主要原因之一。为了获得安全健康的生活环境、减少细菌对人体的伤害,人类不断探索开发环保、安全、卫生、高效和廉价的抗菌材料及其在产品中的应用。目前,用于抑菌或消灭有害微生物的杀菌剂根据其杀菌成分的化学结构主要分为三大类: 天然抗菌剂、无机抗菌剂和有机抗菌剂。天然抗菌剂主要为天然植物的提取物,受资源的限制, 应用推广有一定困难; 无机抗菌剂这类化合物的耐洗性很差, 杀菌作用比较慢; 有机抗菌剂主要包括醛类、酚类、醇类、过氧化物类、胍类、醚类等。
3.双胍盐属于胍类有机抗菌剂,具有优良的杀菌抑菌作用和良好的广谱杀菌性,其抗菌机理是:双胍盐的正电荷中心[n
],使细胞膜中酶的代谢功能失活,使其呼吸功能受到破坏;可通过渗透扩散吸附作用,吸附带负电荷的微生物,并破坏微生物的细胞结构和物质能量代谢,从而起到杀菌灭菌的作用。
[0004]
双胍盐类抗菌剂中最优异的一种是英国avecia公司开发的聚已亚甲基盐酸(phmb)抗菌剂,其合成路线是由己二胺和双氰胺的铜盐或锌盐在60℃加热反应,通二氧化氯使ph值由11降至6.8-7,冷却过滤,先制得己亚甲基二胺的二氰酸盐,再用己二胺和36%的盐酸处理,然后加热至150-155℃,搅拌反应4小时,得产品。由于混杂有各种反应中间体及副产物,要制得高纯度的产物非常困难,难以满足一些对高纯度产品的需求,同时这种抗菌材料的抗菌性能也不够持久, 将其用于织物上所制得的抗菌织物在100次洗涤后基本上就不再具备抗菌性。如何通过简单廉价的方法制得高纯度的双胍盐类抗菌剂材料,以及如何进一步提高双胍盐类抗菌剂材料的抗菌性和耐久性将是非常具有挑战性的课题。
[0005]
鉴于现有技术存在的问题,需要对传统的抗菌材料的制备方法和结构进行改造以解决现有技术存在的问题,以期实现合成路线短、合成工艺简单、原料成本低、环境污染小,更易实现规模化生产等特性,具有重要意义。
技术实现要素:
[0006]
鉴于上述现有技术中存在的问题,本发明开发了一种有机硅季铵盐双胍盐材料及其制备方法,其制备方法工艺简单,原料价廉易得,有效解决了现在使用的双胍盐材料在合成技术上存在的问题,所获产物克服了现有抗菌产品在使用性能上存在的不足,具有广泛的应用范围。
[0007]
本发明的目的之一是提供一种有机硅季铵盐双胍盐材料,化学结构如6所示,
式中,x1选自氯、溴、碘、对甲苯磺酰氧基、苯磺酰氧基、甲磺酰氧基、乙酰氧基、葡萄糖酸根、乳酸根、羟基乙酸根;x2选自氯、溴、碘、对甲苯磺酰氧基、苯磺酰氧基、甲磺酰氧基;r1选自甲基、乙基、c3-c8烷基、苯基、4-氯苯基、4-溴苯基、4-氟苯基、4-碘苯基、4-三氟甲氧基苯基、4-甲氧基苯基、4-乙氧基苯基、4-甲基苯基、4-三氟甲基苯基;r2选自甲基、乙基、c3-c6烷基、苯基、乙烯基、烯丙基,l=1-100, m=1-100,n=1-100。
[0008]
在本发明的一较佳实施例中,上述的l,m和n满足的和大于或等于3,且小于或等于40。
[0009]
本发明的目的之二是提供一种抗菌组合物,按照质量百分比计,包括上述有机硅季铵盐双胍盐材料1-3份,有机硅偶联抗菌剂1-5份和水97-99份。
[0010]
在本发明的一较佳实施例中,上述的有机硅偶联抗菌剂为二甲基十六烷基[3-(三甲氧基硅基)丙基]氯化铵、二甲基十六烷基[3-(三乙氧基硅基)丙基]氯化铵、二甲基十八烷基[3-(三甲氧基硅基)丙基]氯化铵、二甲基十八烷基[3-(三乙氧基硅基)丙基]氯化铵、二甲基二十烷基[3-(三甲氧基硅基)丙基]氯化铵、二甲基二十烷基[3-(三乙氧基硅基)丙基]氯化铵、二甲基十六烷基[3-(三甲氧基硅基)丙基]溴化铵、二甲基十六烷基[3-(三乙氧基硅基)丙基] 溴化铵、二甲基十八烷基[3-(三甲氧基硅基)丙基] 溴化铵、二甲基十八烷基[3-(三乙氧基硅基)丙基] 溴化铵、二甲基二十烷基[3-(三甲氧基硅基)丙基] 溴化铵、二甲基二十烷基[3-(三乙氧基硅基)丙基] 溴化铵、二甲基十六烷基[3-(三甲氧基硅基)丙基]碘化铵、二甲基十六烷基[3-(三乙氧基硅基)丙基] 碘化铵、二甲基十八烷基[3-(三甲氧基硅基)丙基] 碘化铵、二甲基十八烷基[3-(三乙氧基硅基)丙基] 碘化铵、二甲基二十烷基[3-(三甲氧基硅基)丙基] 碘化铵、二甲基二十烷基[3-(三乙氧基硅基)丙基] 碘化铵中的至少一种。
[0011]
本发明的目的之三是提供一种有机硅季铵盐双胍盐材料的制备方法, 包括如下步骤:
步骤(a)在隔绝空气情况下,在反应容器中,将式1化合物和二氰胺钠加入第一反应溶剂中反应,在加入酸性物质的酸性环境中进行缩合反应,反应温度为60-200℃,反应时间为12-72小时,反应液冷却,后处理制得所述的式2化合物;步骤(b)所述的式2化合物和式3化合物含取代基的伯胺在第二反应溶剂中,酸性条件下反应,反应温度为60-200℃,反应时间为12-72小时,得所述的式4化合物;步骤(c)在隔绝空气情况下,将式4化合物和式5化合物在第三反应溶剂中发生反应,反应温度为100-200℃,反应时间为10-200小时,得化学结构如6所示有机硅季铵盐双胍盐材料;其中,x1选自氯、溴、碘、对甲苯磺酰氧基、苯磺酰氧基、甲磺酰氧基、乙酰氧基、葡萄糖酸根、乳酸根、羟基乙酸根;x2选自氯、溴、碘、对甲苯磺酰氧基、苯磺酰氧基、甲磺酰氧基;r1选自甲基、乙基、c3-c8烷基、苯基、4-氯苯基、4-溴苯基、4-氟苯基、4-碘苯基、4-三氟甲氧基苯基、4-甲氧基苯基、4-乙氧基苯基、4-甲基苯基、4-三氟甲基苯基;r2选自甲基、乙基、c3-c6烷基、苯基、乙烯基、烯丙基,l=1-100, m=1-100,n=1-100。
[0012]
在本发明的一较佳实施例中,上述第一反应溶剂为正丁醇,乙醇,甲醇,异丙醇、丙二醇、乙二醇、乙二醇二乙醚,乙二醇二甲醚,dmf,dmso,乙腈,四氢呋喃,2-甲基四氢呋喃,二氧六环中的至少一种。
[0013]
在本发明的一较佳实施例中,上述第二反应溶剂为乙醇,甲醇,异丙醇、正丁醇、丙二醇、乙二醇、乙二醇二乙醚,乙二醇二甲醚,dmf,dmso,乙腈,四氢呋喃,2-甲基四氢呋喃,二氧六环中的至少一种。
[0014]
在本发明的一较佳实施例中,上述第三反应溶剂为二甲苯,对二甲苯,甲苯,乙二醇二乙醚, dmf,dmso中的至少一种。
[0015]
在本发明的一较佳实施例中,上述的l,m和n满足的和大于或等于3,且小于或等于40。
[0016]
本发明的目的之四是提供一种抗菌织物,包括基底层和抗菌层,上述的所述的抗
菌层包括上述的有机硅季铵盐双胍盐材料或者上述的抗菌组合物或者如上述方法制得的有机硅季铵盐双胍盐材料。
[0017]
有益效果(1)本发明的有机硅季铵盐双胍盐材料同时引入双胍基结构、季铵盐结构和有机硅结构,其中有机硅结构,季铵盐结构和双胍基结构的协同作用,使其抗菌效果比相应的双胍基抗菌材料的抗菌效果有了5倍以上的提升。
[0018]
(2)本发明抗菌广谱,使用本发明的有机硅季铵盐双胍盐抗菌材料对棉质织物进行抗菌整理后所制得的抗菌织物具有非常优异的抗菌性能,对常见菌种(金黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922,白色念球菌atcc 10231,黑曲霉菌atcc 16404)的抑菌效果都很好,抑菌率均有99.9%。
[0019]
(3)本发明耐洗性尤其好,与现有技术相比,50次洗涤后的抑菌率都保持在99.9%,100次洗涤后的抑菌率都在70%以上,200次洗涤后的抑菌率也都还在50%以上。
[0020]
(4)与现有技术相比,本发明的制备方法工艺简单,原料价廉易得,同时具有高转化率,低成本,高安全性,更易工业化放大生产。
具体实施方式
[0021]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述地实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围.下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0022]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0023]
实施例1实施例1
步骤(a):式2-1化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l 正丁醇,1.0 mol式1-1化合物和3.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至110℃反应。保温反应20小时,反应完成。
[0024]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到0.913mol式2-1化合物白色固体产品。
[0025]
收率91.3%,产品滴定纯度97.8%。
[0026]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.51-3.68(m,24h)。
[0027]
步骤(b):式4-1化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l 乙醇,0.913 mol式2-1化合物和3.0 mol式 3-1化合物正己胺乳酸盐,加完后,搅匀;反应液升温至78 ℃反应。保温反应56小时,反应完成。
[0028]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.822mol式4-1化合物白色固体产品。
[0029]
收率90.0%,产品滴定纯度97.1%。
[0030]
核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.85(t,9h),1.21-1.26(t,18h),1.31(d,9h),1.36(m,6h),2.68(t,6h),3.02(t,6h),3.48(t,6h),3.51-3.68(m,24h) ,4.13(q,3h)。
[0031]
步骤(c):式6-1化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l 二甲苯,0.822 mol式4-1化合物,1.0 mol式5-1化合物氯丙基三甲氧基硅烷,加完后,搅匀;反应液升温至135 ℃反应。保温反应35小时,反应完成。
[0032]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.776 mol式6-1化合物白色固体产品。
[0033]
收率94.4%,三步反应总收率77.6%,产品滴定纯度:98.5%核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.61(t,2h),0.85(t,9h),1.21-1.26(t,18h),1.31(d,9h),1.36(m,6h),1.72(m,2h),3.02(t,6h),3.28(t,6h),3.36(t,2h),3.48(t,6h),3.51-3.65(m,33h) ,4.13(q,3h)。
[0034]
实施例2实施例2实施例2步骤(a):式2-2化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l甲醇,1.0 mol式1-2化合物和4.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至60 ℃反应。保温反应72小时,反应完成。
[0035]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到0.924mol式2-2化合物白色固体产品。
[0036]
收率92.4%,产品滴定纯度98.4%。
[0037]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.57(t,6h),3.65(t,6h)。
[0038]
步骤(b):式4-2化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l异丙醇,0.924 mol式2-2化合物和5.0 mol式3-2化合物对氯苯胺盐酸盐,加完后,搅匀;反应液升温至80℃反应。保温反应50小时,反应完成。
[0039]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.841mol式4-2化合物白色固体产品。
[0040]
收率91.0%,产品滴定纯度97.2%。
[0041]
核磁数据:1h nmr (400mhz,dmso-d6/d2o)):δ2.68(t,6h),3.48(t,6h),3.57(t,
6h),3.65(t,6h) ,7.02(d,6h) ,7.11(d,6h)。
[0042]
步骤(c):式6-2化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l对二甲苯,0.841mol式4-2化合物,2.0 mol式5-2化合物溴丙基三乙氧基硅烷,加完后,搅匀;反应液升温至138 ℃反应。保温反应30小时,反应完成。
[0043]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.765 mol式6-2化合物白色固体产品。
[0044]
收率91%,三步反应总收率76.5%,产品滴定纯度:98.1%。
[0045]
核磁数据:1h nmr (400mhz,dmso-d6/d2o)):δ0.61(t,2h),1.19(t,9h),1.72(m,2h), 3.28(t,6h),3.36(m,2h),3.48(t,6h),3.61-3.69(m,12h) ,3.81(q,6h) ,7.02(d,6h) ,7.11(d,6h)。
[0046]
实施例3实施例3实施例3步骤(a):式2-3化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l丙二醇,1.0 mol式1-3化合物和5.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至180 ℃反应。保温反应10小时,反应完成。
[0047]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行
萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到0.908mol式2-3化合物白色固体产品。
[0048]
收率90.8%,产品滴定纯度97.6%。
[0049]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.51-3.72(m,24h)。
[0050]
步骤(b):式4-3化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l丙二醇,0.908 mol式2-3化合物和6.0 mol式3-3化合物正丁胺乙酸盐,加完后,搅匀;反应液升温至180 ℃反应。保温反应10小时,反应完成。
[0051]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.835mol式4-3化合物白色固体产品。
[0052]
收率92.0%,产品滴定纯度98.1%。
[0053]
核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.87(t,9h),1.24(m,6h),1.34(m,6h),1.93(s,9h),2.68(t,6h),3.02(t,6h),3.48(t,6h),3.51-3.72(m,24h)。
[0054]
步骤(c):式6-3化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l甲苯,0.835 mol式4-3化合物,4.0 mol式5-3化合物溴丙基三异丙氧基硅烷,加完后,搅匀;反应液升温至110 ℃反应。保温反应45小时,反应完成。
[0055]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.756 mol式6-3化合物白色固体产品。
[0056]
收率90.5%,两步反应总收率77.6%,产品滴定纯度:98.2%。
[0057]
核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.61(t,2h),0.87(t,9h),1.20(d,18h),1.24(m,6h),1.34(m,6h),1.72(m,2h),1.93(s,9h),3.28(t,6h),3.02(t,6h),3.36(m,2h),3.48(t,6h),3.51-3.72(m,24h) ,3.79(m,3h)。
[0058]
实施例4实施例4
步骤(a):式2-4化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l乙二醇二乙醚,1.0 mol式1-4化合物和6.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至120 ℃反应。保温反应18小时,反应完成。
[0059]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到0.897mol式2-4化合物白色固体产品。
[0060]
收率89.7%,产品滴定纯度98.6%。
[0061]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.51-3.76(m,108h)。
[0062]
步骤(b):式4-4化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l乙二醇二乙醚,0.897 mol式2-4化合物和7.0 mol式3-4化合物苯胺氢溴酸盐,加完后,搅匀;反应液升温至120 ℃反应。保温反应20小时,反应完成。
[0063]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.798mol式4-4化合物白色固体产品。
[0064]
收率89.0%,产品滴定纯度98.2%。
[0065]
核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ2.68(t,6h),3.48(t,6h),3.51-3.76(m,108h) ,6.29-7.13 (m,9h) ,7.62 (d,6h)。
[0066]
步骤(c):式6-4化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l乙二醇二乙醚,0.798mol式4-4化合物,6.0 mol式5-4化合物碘丙基三乙氧基硅烷,加完后,搅匀;反应液升温至120 ℃反应。保温反应40小时,反应完成。
[0067]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.753 mol式6-4化合物白色固体产品。
[0068]
收率94.4%,两步反应总收率75.3%,产品滴定纯度:97.7%核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.61(t,2h),1.19(t,9h),1.72(m,2h),3.28(t,6h),3.36(m,2h),3.48(t,6h),3.51-3.76(m,108h) ,3.81(q,6h) ,6.29-7.13 (m,9h) ,7.62 (d,6h)。
[0069]
实施例5
步骤(a):式2-5化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l dmf,1.0 mol式1-5化合物和7.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至150℃反应。保温反应15小时,反应完成。
[0070]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到0.922mol式2-5化合物白色固体产品。
[0071]
收率92.2%,产品滴定纯度96.6%。
[0072]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.51-3.78(m,160h)。
[0073]
步骤(b):式4-5化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l 乙醇,0.922mol式2-5化合物和8.0 mol甲胺羟基乙酸盐,加完后,搅匀;反应液升温至78 ℃反应。保温反应56小时,反应完成。
[0074]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.812mol式4-5化合物白色固体产品。
[0075]
收率88.1%,产品滴定纯度97.5%。
[0076]
核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ2.68(t,6h),2.73(s,9h),3.48(t,6h),3.51-3.78(m,160h) ,3.95(s,6h)。
[0077]
步骤(c):式6-5化合物的制备
保持氮气微正压,向20 l反应瓶中依次加入5l乙二醇二乙醚,,0.812 mol式4-5化合物,7.0 mol式5-5化合物氯丙基三苯氧基硅烷,加完后,搅匀;反应液升温至120 ℃反应。保温反应41小时,反应完成。
[0078]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.723mol式6-5化合物白色固体产品。
[0079]
收率89.0%,两步反应总收率72.3%,产品滴定纯度:97.7%核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.61(t,2h),1.72(m,2h),2.73(s,9h),3.28(t,6h),3.36(m,2h),3.48(t,6h), 3.51-3.77(m,160h) ,3.95(s,6h) ,6.81-7.22(m,15h)。
[0080]
实施例6实施例6实施例6步骤(a):式2-6化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l四氢呋喃,1.0 mol式1-6化合物和8.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至66 ℃反应。保温反应60小时,反应完成。
[0081]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到式2-6化合物0.887mol白色固体产品。
[0082]
收率88.7%,产品滴定纯度97.7%。
[0083]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.51-3.83(m,240h)。
[0084]
步骤(b):式4-6化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l四氢呋喃,0.887mol式2-6化合物和式3-6化合物10.0 mol对三氟甲氧基苯胺对甲苯磺酸盐,加完后,搅匀;反应液升温至66 ℃反应。保温反应65小时,反应完成。
[0085]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.816mol式4-6化合物白色固体产品。
[0086]
收率92.0%,产品滴定纯度98.3%。
[0087]
核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ2.29(s,9h),2.68(t,6h),3.45(t,6h),3.51-3.82(m,240h),7.17(d,6h),7.31(d,6h),7.42(d,6h),7.53(d,6h)。
[0088]
步骤(c):式6-6化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l dmf,0.816 mol式4-6化合物,9.0 mol式5-6化合物溴丙基三苄氧基硅烷,加完后,搅匀;反应液升温至150 ℃反应。保温反应28小时,反应完成。
[0089]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.726 mol式6
ꢀ‑
6化合物白色固体产品。
[0090]
收率89.0%,两步反应总收率72.6%,产品滴定纯度:98.4%核磁数据:1h nmr (400mhz,dmso-d6/d2o):δ0.61(t,2h),1.72(m,2h),2.29(s,9h),3.28(t,6h),3.36(m,2h),3.45(t,6h),3.51-3.82(m,240h),5.06(s,6h),7.17(d,6h),7.24-7.46(m,27h),7.53(d,6h)。
[0091]
实施例7实施例7
步骤(a):式2-7化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l二氧六环,1.0 mol式1-7化合物和10.0 mol二氰胺钠,加完后,搅匀;再向其中滴加500 ml浓盐酸,加完后,反应液升温至101 ℃反应。保温反应25小时,反应完成。
[0092]
反应液冷却至室温,向其中加入氢氧化钠,调ph至9-10。向其中加入乙酸乙酯进行萃取。萃取液旋干,再用适量乙酸乙酯-石油醚混合溶液进行重结晶,析出大量白色固体,过滤,收集滤饼得到0.935mol式2-7化合物白色固体产品。
[0093]
收率93.5%,产品滴定纯度97.8%。
[0094]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.65(t,6h),3.45(t,6h),3.50-3.84(m,440h)。
[0095]
步骤(b):式4-7化合物的制备保持氮气微正压,向20 l反应瓶中依次加入3l二氧六环,0.935 mol式2-7化合物和3.0 mol式3-7化合物对甲基苯胺甲磺酸盐,加完后,搅匀;反应液升温至101℃反应。保温反应25小时,反应完成。
[0096]
反应液冷却至室温,向其中加入适量叔丁基甲醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.853mol式4-7化合物白色固体产品。
[0097]
收率91.2%,产品滴定纯度97.5%。
[0098]
核磁数据:1h nmr (400mhz,dmso-d6):δ2.51(s,9h),2.68(t,6h),2.87(s,9h),3.49(t,6h),3.50-3.83(m,440h),6.82(d,6h) ,7.17(d,6h)。
[0099]
步骤(c):式6-7化合物的制备保持氮气微正压,向20 l反应瓶中依次加入5l dmso,0.853 mol式4-7化合物,10.0 mol式5-7化合物对甲苯磺酰氧丙基三烯丙氧基硅烷,加完后,搅匀;反应液升温至180℃反应。保温反应15小时,反应完成。
[0100]
反应液冷却至室温,向其中加入适量石油醚,冷却至-20℃,析出大量固体,过滤,收集滤饼得到0.785 mol式6-7化合物白色固体产品。
[0101]
收率92.0%,两步反应总收率78.5%,产品滴定纯度:98.1%。
[0102]
核磁数据:1h nmr (400mhz,dmso-d6):δ0.61(t,2h),1.72(m,2h),2.29(s,3h),2.51(s,9h),2.65(t,6h),2.87(s,9h),3.28(t,6h),3.36(m,2h),3.49(t,6h),3.50-3.84(m,440h),4.15(m,6h),5.16(m,6h),5.29(m,6h),5.99(m,6h),6.82(d,6h),7.13-7.17(m,8h) ,6.53(d,2h)。
[0103]
化合物抗菌性能测试:mic(最低抑制浓度)的测定:采用微量肉汤稀释法将实施例1-7制得的化合物(4-1,4-2,4-3,4-4,4-5,4-6,4-7,6-1,6-2,6-3,6-4,6-5,6-6,6-7)混合在lb营养肉汤中做系列的二倍稀释,加入定量的受试菌经过一定时间培养后,观察到无细菌生长的最低化合物浓度即为该化合物对此菌的mic(最低抑制浓度)。
[0104]
具体测定步骤如下:(1)悬菌液的制备:在无菌操作台上,用灭菌的接种环挑取适量细菌培养物,移种至10mllb肉汤培养液中,在37℃摇床中培养6-8h,以待菌液至轻微或中度浊度。为保证药敏试验的准确度和精确度,必须对接种菌液的浓度做相应控制。因此,移取少量菌液于比色管中,稀释至0.5麦氏标准浓度后稀释1000倍,菌液含量约为1
×
105cfu/ml。
[0105]
(2)抗菌化合物母液的制备:将化合物溶于无菌水中,制备成特定浓度的抗菌化合物母液,并用无菌过滤头除去溶液可能含有的细菌。
[0106]
(3)mic平板的制备:96孔板第2列至第10列的第2行至第7行每孔加入100μllb肉汤,第2列加100μl抗菌化合物母液,用移液枪吹打混匀后吸取100μl至第3列,以此类推,共8个浓度梯度,第9列弃去100μl的混合液,第10列不加药液做阳性对照,然后每孔加入100μl悬菌液,将混合液用移液枪吹打均匀。第11列不加菌液加入200μllb肉汤做阴性对照。化合物和菌液吹打混合完毕后,盖上96孔板盖,置于37℃生化培养箱内培养,培养20-24h(大肠杆菌atcc25922、金黄色葡萄球菌atcc6538)或28℃生化培养箱内培养40-48h(白色念球菌atcc10231、黑曲霉菌atcc16404),用酶标仪测定菌液的od570值(光密度)。
[0107]
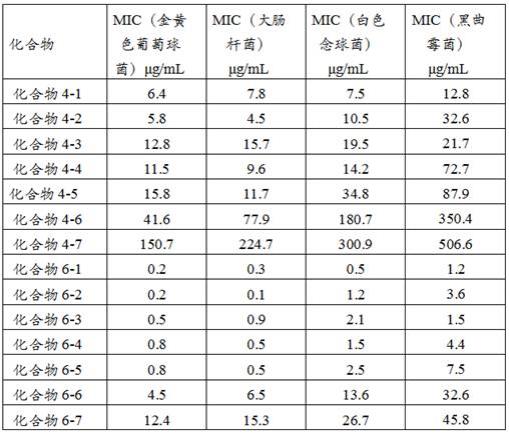
(4)判定结果:mic(最低抑制浓度)为在96孔板内完全抑制细菌生长的浓度,具体结果如表1所示。
[0108]
表1实施例1-7中化合物对部分微生物的最低抑制浓度(mic)
通过上述实验结果可以看出,有机硅季铵盐双胍盐材料(化合物6-1、化合物6-2、化合物6-3、化合物6-4、化合物6-5、化合物6-6、化合物6-7) 对常见菌种(金黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922,白色念球菌atcc 10231,黑曲霉菌atcc 16404)的抑菌效果都很优异,最低抑菌浓度都在100μg/ml以下;尤其是当化合物分子式中l,m和n满足的和大于或等于3,且小于或等于40时,抑菌效果尤其优异,其最低抑菌浓度都在10μg/ml以下,效果最好的甚至低至0.1μg/ml。
[0109]
同时,制备有机硅季铵盐双胍盐材料的中间体化合物(化合物4-1、化合物4-2、化合物4-3、化合物4-4、化合物4-5、化合物4-6、化合物4-7)也表现出了很好的抑菌效果,对常见菌种的最低抑菌浓度也都在1000μg/ml以下,但总体来说,都要比相应的有机硅季铵盐双胍盐材料差不少。通过与有机硅试剂反应引入有机硅基团和季铵盐基团,取得了意料之外的技术效果,使抗菌效果有了5倍以上的提升。按照行业里的普遍认知,单个双胍盐基团的抗菌活性会略高于或相当于季铵盐基团,如果在3个双胍盐基团的基础上加上1个抗菌活性相当或较弱的季铵盐基团和1个明显无抗菌活性的有机硅基团,其总体抗菌活性应该会只是比3个双胍盐基团的抗菌活性提高50%以下,而本发明则发现其总体抗菌活性提升了5倍以上,非常意外。造成这一意外技术效果的原因暂时还不明了,还在继续研究中。
[0110]
化合物在织物抗菌整理上的性能测试:按照质量百分比计,包括将实施例1-7制得的化合物(4-1,4-2,4-3,4-4,4-5,4-6,4-7,6-1,6-2,6-3,6-4,6-5,6-6,6-7)料2份, 有机硅偶联抗菌剂3份和水98份,制得抗菌组合物8-21。
[0111]
其中,在抗菌组合物 8中,有机硅偶联抗菌剂为二甲基十六烷基[3-(三甲氧基硅
基)丙基]氯化铵。
[0112]
在抗菌组合物9中,有机硅偶联抗菌剂为二甲基十六烷基[3-(三甲氧基硅基)丙基]氯化铵。
[0113]
在抗菌组合物10中,有机硅偶联抗菌剂为二甲基十六烷基[3-(三乙氧基硅基)丙基]氯化铵。
[0114]
在抗菌组合物11中,有机硅偶联抗菌剂为二甲基十六烷基[3-(三乙氧基硅基)丙基]氯化铵。
[0115]
在抗菌组合物12中,有机硅偶联抗菌剂为二甲基十八烷基[3-(三乙氧基硅基)丙基]氯化铵。
[0116]
在抗菌组合物13中,有机硅偶联抗菌剂为二甲基十八烷基[3-(三乙氧基硅基)丙基]氯化铵。。
[0117]
在抗菌组合物14中,有机硅偶联抗菌剂为二甲基二十烷基[3-(三甲氧基硅基)丙基]氯化铵。
[0118]
在抗菌组合物15中,有机硅偶联抗菌剂为二甲基二十烷基[3-(三甲氧基硅基)丙基]氯化铵。
[0119]
在抗菌组合物16中,有机硅偶联抗菌剂为二甲基二十烷基[3-(三乙氧基硅基)丙基]氯化铵。
[0120]
在抗菌组合物17中,有机硅偶联抗菌剂为二甲基二十烷基[3-(三乙氧基硅基)丙基]氯化铵。
[0121]
在抗菌组合物18中,有机硅偶联抗菌剂为二甲基十六烷基[3-(三甲氧基硅基)丙基]溴化铵。
[0122]
在抗菌组合物19中,有机硅偶联抗菌剂为二甲基十六烷基[3-(三甲氧基硅基)丙基]溴化铵。
[0123]
在抗菌组合物20中,有机硅偶联抗菌剂为二甲基二十烷基[3-(三乙氧基硅基)丙基] 碘化铵。
[0124]
在抗菌组合物21中,有机硅偶联抗菌剂为二甲基二十烷基[3-(三乙氧基硅基)丙基] 碘化铵。
[0125]
将抗菌组合物8-21,浴比1:15,将纯棉织物放入该抗菌整理剂溶液中浸泡10分钟,然后通过压辊,轧余率80%,再将织物放入150℃烘房焙烘5分钟,将织物从烘房取出,分别得到相应的抗菌织物。
[0126]
抗菌织物抗菌性测试: 参考gb/120944.3-2008《纺织品抗菌性能的评价第3部分:振荡法》,菌种选择金黄色葡萄球菌(atcc 6538),大肠杆菌(atcc 25922),白色念球菌(atcc 10231),黑曲霉菌(atcc 16404),具体结果如表2所示。
[0127]
表2 实施例1-7中化合物所制得抗菌织物的抑菌率
ꢀ
从上述实验结果可以看出,使用本发明的有机硅季铵盐双胍盐抗菌材料对棉质织物进行抗菌整理后所制得的抗菌织物具有非常优异的抗菌性能,对常见菌种(金黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922,白色念球菌atcc 10231,黑曲霉菌atcc 16404)的抑菌效果都很好,抑菌率均有99.9%。并且,这些抗菌织物都具有良好的耐洗性,都能耐洗涤,50次洗涤后的抑菌率都在90%以上。更优异的是,当化合物分子式中l,m和n满足的和大于或等于3,且小于或等于40时,耐洗性尤其好,50次洗涤后的抑菌率都保持在99.9%,100次洗涤后的抑菌率都在70%以上,200次洗涤后的抑菌率也都还在50%以上。
[0128]
同时,使用本发明的中间体化合物对棉质织物进行抗菌整理后所制得的抗菌织物也都具有非常优异的抗菌性能,对常见菌种(金黄色葡萄球菌atcc 6538,大肠杆菌atcc 25922,白色念球菌atcc 10231,黑曲霉菌atcc 16404)的抑菌效果都很好,抑菌率均有99.9%。不过,这些抗菌织物的耐洗性会相对来说要差一些,100次和200次洗涤后都已经基本上没有了抗菌性,50次洗涤后大部分抗菌织物的抑菌率也有较大幅度的下降。由此可见,
通过与有机硅试剂反应同时引入有机硅基团和季铵盐基团,使有机硅季铵盐双胍盐抗菌材料的耐洗性有了显著提升。
[0129]
上述实例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人是能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围;凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。