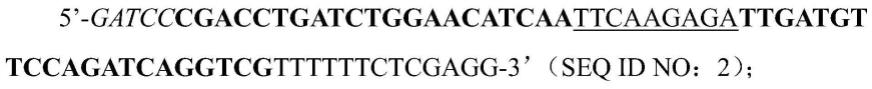
parp-1shrna重组慢病毒载体、该载体稳定转染hacat的细胞系及其应用
技术领域
1.本发明属于分子生物学和基因工程技术领域,涉及parp-1 shrna重组慢病毒载体、该载体稳定转染hacat的细胞系及其应用。
背景技术:
2.皮肤癌是人类中最常见的癌症之一,是发生于皮肤的恶性肿瘤,根据发生部位的不同,分为鳞状细胞癌(squamous cell carcinoma)、基底细胞癌(basal cell carcinoma,bcc)、恶性黑色素瘤(malignant melanoma)、恶性淋巴瘤 (malignant lymphoma,ml)等
[1,2]
。暴露于太阳辐射是皮肤癌最广为人知的诱因
[3]
。另外,流行病学研究表明,其他类型的辐射(例如,电离辐射)、农药、空气污染中的颗粒物、水和某些食物中的毒素等环境污染物造成的dna 损伤也被认为是导致皮肤癌的重要因素
[4]
。
[0003]
皮肤细胞基因组损伤后会激活多种dna损伤修复酶
[5]
,其中包括最重要的dna损伤修复酶之一:聚腺苷二磷酸核糖聚合酶-1(poly(adp-ribose) polymerase,parp-1)
[6]
。聚腺苷二磷酸核糖基化(poly(adp-ribosyl)ation, parylation)是一种可在生理及病理条件下发生的翻译后修饰,主要参与到遗传毒性的应激反应中。parp-1可以结合到dna单链或双链损伤处,通过催化底物烟酰胺腺嘌呤二核苷酸(nad )合成长达200个adp-ribose单位以上聚腺苷二磷酸核糖(poly(adp-ribose),par),产生parylation的作用
[7]
。 parylation作用是指par通过共价或非共价的方式结合到靶蛋白上,对靶蛋白进行修饰的过程。通过这种修饰,par可以调节靶蛋白的理化性质,从而介导染色质重塑、dna损伤修复、转录、细胞内信号传导、细胞周期和死亡、表观遗传学调控等多种细胞功能
[8]
。目前,parylation与dna损伤修复相关的研究较多,parp家族共有17个亚型,其中parp-1作为含量最多的亚型,贡献了机体90%的parylation的作用,parp-1作为重要的dna损伤修复酶被广泛关注
[9]
。
[0004]
目前皮肤癌防治药物筛选的细胞模型主要为癌细胞筛选模型,而辐射及环境污染物主要损伤的是正常皮肤细胞的dna。
[0005]
rna干扰技术是抑制基因表达的常规分子生物学手段,其原理是当细胞中导入与内源性mrna编码区同源的双链rna时,该mrna降解而导致基因表达沉默的现象。
[0006]
基因治疗的载体分为非病毒载体和病毒载体,其中非病毒载体无法满足长时间的表达,这一缺陷被病毒载体填补。近年来慢病毒介导的rna干扰技术在研究中广泛引用。
[0007]
慢病毒载体(lentivirus-based vector,lv)是逆转录病毒的一种,其具有逆转录病毒的基本结构,但又存在不同于逆转录病毒的组分特性,该病毒既可以转染分裂细胞又可以转染非分裂细胞。病毒基因组可以整合在宿主细胞中,使得基因能够长时间稳定表达,并且慢病毒具有较低的免疫源性,使得慢病毒成为rna干扰的最佳载体。
[0008]
本研究以人永生化表皮角化形成细胞(hacat)为基础,在hacat细胞中稳定转染parp-1的短发夹rna(short hairpin rna,shrna)并筛选单克隆稳转细胞系,填补了正常皮肤细胞dna损伤防治药物筛选模型的空白,为寻找dna损伤防治药物的相关研究奠定基础。
技术实现要素:
[0009]
发明要解决的问题
[0010]
针对现有技术中存在的问题,本技术提供了一种parp-1 shrna重组慢病毒载体、重组慢病毒载体稳定转染hacat的细胞系及其制备方法。
[0011]
用于解决问题的方案
[0012]
本发明人鉴于上述现有技术中存在的问题,进行了深入的研究、反复试验,针对parp-1mrna设计干扰序列shrna,并将其插入到双酶切后的慢病毒载体质粒中得到重组慢病毒载体;进而将该重组慢病载体转染至hacat 细胞,并筛选单克隆细胞系,从而完成了本发明。即本发明如下所述:
[0013]
在本发明的第一方面,提供了一种重组慢病毒载体,其特征在于,所述重组慢病毒载体是在plvx-shrna2-puro载体质粒的多克隆位点中插入了 parp-1 shrna片段,其中所述parp-1 shrna片段中含有seq id no:1所示的序列。
[0014]
在优选的实施方式中,所述parp-1 shrna片段由seq id no:2所示的上游序列片段和seq id no:3所示的下游序列片段退火后形成。
[0015]
在本发明的第二方面,提供了本发明的第一方面中所述重组慢病毒载体的制备方法,其包括以下步骤:
[0016]
(1)以seq id no:1所示的序列为基础,合成parp-1 shrna片段的上游序列片段和下游序列片段;优选的,所述上游序列片段的核苷酸序列如 seq id no:2所示,所述下游序列片段的核苷酸序列如seq id no:3所示;
[0017]
(2)对plvx-shrna2-puro载体质粒用ecor i bamh i进行双酶切;
[0018]
(3)将步骤(1)中所述的上游序列片段和下游序列片段与步骤(2) 中经过双酶切后的载体质粒进行连接;优选的,连接条件为22℃水浴过夜。
[0019]
在本发明的第三方面,提供了一种慢病毒颗粒,其特征在于,所述慢病毒颗粒是通过将本发明的第一方面中所述的重组慢病毒载体与包装质粒共转染至hek293t细胞中,并将所述细胞在完全培养基中进行培养后收集得到的;优选的,所述包装质粒为pspax2和pmd2.g的混合物,所述完全培养基为dmem 10%fbs。
[0020]
在本发明的第四方面,提供了一种稳定表达parp-1 shrna的hacat细胞系,其特征在于,所述细胞系是通过将本发明的第三方面中所述的慢病毒颗粒转染至hacat细胞中制备得到的;优选的,所述稳定表达parp-1 shrna 的hacat细胞系为单克隆细胞系。
[0021]
在本发明的第五方面,提供了一种制备稳定表达parp-1 shrna的 hacat细胞系的方法,其包括以下步骤:
[0022]
(1)将本发明的第三方面所述的慢病毒颗粒添加至hacat细胞中,其中所述慢病毒颗粒与所述hacat细胞之间的数量比为100:1~1:1;
[0023]
(2)20~28小时后,在培养基中加入1~6μg/ml的嘌呤霉素进行筛选;
[0024]
(3)筛选4~7天后,将所述细胞转移至新的培养板中,待细胞长满后转移至培养瓶中进行扩大培养,从而得到稳定表达parp-1 shrna的hacat细胞系。
[0025]
在优选的实施方式中,步骤(1)中所述慢病毒颗粒与所述hacat细胞之间的数量比为100:1,步骤(2)中所述嘌呤霉素的浓度为3μg/ml。
[0026]
在另一个优选的实施方式中,上述方法还进一步包括以下步骤:
[0027]
(4)通过极限稀释法,将步骤(3)中所述细胞系重新接种到96孔板中,每孔接种量为100μl、细胞悬液浓度为1个细胞/100μl;待细胞贴壁生长后,置于显微镜下观察,标记至含1个细胞的培养孔,培养12~18天后,得到稳定表达parp-1 shrna的单克隆hacat细胞系;优选的,细胞培养的时间为15 天。
[0028]
在本发明的第六方面,提供了利用本发明的第四方面中所述的稳定表达 parp-1 shrna的hacat细胞系在筛选预防或治疗皮肤癌的药物中的应用。
[0029]
在本发明的第七方面,提供了利用本发明的第四方面中所述的稳定表达 parp-1 shrna的hacat细胞系在筛选预防或治疗正常皮肤细胞dna损伤的药物中的应用。
[0030]
发明的效果
[0031]
由本发明的技术方案可见,本发明的技术方案与现有技术相比,具有以下有益效果:
[0032]
1,成功构建parp-1 shrna的慢病毒载体,并将包装后的慢病毒转染至 hacat细胞,获得4株稳定表达parp-1 shrna的单克隆细胞系。
[0033]
2,western blot结果显示本发明构建的单克隆细胞系敲降效率较高。
[0034]
3,dna损伤试验结果表明parp-1敲降显著性增加了dna损伤,提示 parp-1在正常皮肤细胞hacat的dna损伤修复中起到重要作用。
[0035]
4,本发明构建的稳定敲降parp-1的hacat单克隆细胞系,可用于预防或治疗皮肤癌的药物的筛选及正常皮肤细胞dna损伤机制的研究。
[0036]
为了让本发明的上述和其他目的、特征和优点能更明显易懂,下面特举较佳实施例,并配合说明书附图,作详细说明如下:
附图说明
[0037]
图1为带有parp-1 shrna序列的重组阳性克隆的酶切鉴定结果图。其中 m为dna marker;条带1-3均为阳性克隆酶切后的电泳条带。
[0038]
图2为含parp-1 shrna序列的重组质粒的测序结果。
[0039]
图3中的a和b分别为plvx-shrna2-puro-parp-1-kd及包装质粒转染 hek293t细胞在荧光显微镜下的明场图像和绿色荧光蛋白图像(
×
10)。
[0040]
图4示出了不同moi病毒转染hacat细胞后,绿色荧光蛋白(gfp)的表达情况(
×
20)。其中a为对照细胞,b为moi=1,c为moi=10,d为moi=100; a或b后的数字1为明场图像,a或b后的数字2为绿色荧光蛋白图像。
[0041]
图5为不同浓度的嘌呤霉素对hacat细胞毒性作用(
×
20)示意图,其中a为:0μg/ml嘌呤霉素;b为1μg/ml嘌呤霉素;c为3μg/ml嘌呤霉素;d为 6μg/ml嘌呤霉素。
[0042]
图6为荧光显微镜下四株parp-1敲降的hacat单克隆细胞系(
×
20)的明场图像和绿色荧光蛋白图像;a~d分别代表四株单克隆细胞系,其中a或 b后的数字1为明场图像,a或b后的数字2为绿色荧光蛋白图像。
[0043]
图7为western blotting检测nc组细胞与四株parp-1敲降的hacat单克隆细胞系的parp-1蛋白表达情况。
[0044]
图8示出了parp-1敲降对dna损伤的影响情况。其中,a为dna损伤6h 后在未处理组、不同浓度sm处理组的hacat细胞中γ-h2ax的含量;b为 dna损伤24h后在未处理组、不同
浓度sm处理组的hacat细胞中γ-h2ax的含量。
具体实施方式
[0045]
本发明所列举的具体实施例只作为本发明的范例,本发明并不限制于下文所描述的具体实施例。对于本领域技术人员而言,任何对下文所述的实施例进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所有试剂或仪器未注明生产厂商者,均为可以通过市购的常规产品。为了更好地说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在另外一些实施例中,对于本领域技术人员熟知的方法、手段、器材和步骤未作详细描述,以便凸显本发明的主旨。
[0046]
除非另有定义,否则本文中使用的所有技术和科学术语均具有与本领域一般技术人员通常所理解的含义相同的含义。如无特殊说明,本说明书中所使用的单位均为国际标准单位,并且本发明中出现的数值和数值范围,均应当理解为包含了工业生产中所不可避免的系统性误差。
[0047]
仪器、试剂及材料
[0048]
1.仪器
[0049]
细胞计数仪countess(美国invitrogen);ph计f-11(北京屹源电子仪器科技公司);制冰机af100-as(斯科茨曼);荧光显微镜dm4000b(美国leica);台式高速冷冻离心机3k15(美国sigma);生物安全柜bsc-1360(北京东联哈尔);微量紫外分光光度计nd2000c(美国thermo);纯水机milii-q biocel (美国miliipore)。
[0050]
2.试剂及材料
[0051]
jm109感受态细胞(深圳百恩维生物科技有限公司);质粒抽提试剂盒(promega);限制性内切酶ecori(美国neb);限制性内切酶bamhi(美国 neb);限制性内切酶xhoi(美国neb);t4 dna ligase m0202l(美国neb); het高效转染试剂盒bw11002(深圳百恩维生物科技有限公司);dmem培养基(cyclone);胎牛血清fbs(gibco);胰蛋白酶trypsin(上海索莱宝生物科技有限公司);0.22μm针头滤器(millipore);millex-hv 0.45μm pvdf过滤器,货号:slhv-033-rs(millipore);parp-1抗体(cell signaling technology);β-actin抗体(中杉金桥);hrp标记的山羊抗兔或山羊抗鼠二抗(cell signaling technology),western blot发光底物(thermo);蛋白酶抑制剂(roche)。
[0052]
慢病毒过表达载体质粒:plvx-shrna2-puro(百恩维生物科技有限公司);hek293t细胞由本发明人实验室保存,人永生化表皮角化形成细胞 (hacat)购于中国典型培养物保藏中心(china center for type culturecollection,简称cctcc)。
[0053]
实施例1慢病毒载体的构建
[0054]
查阅现有技术文献
[10]
获得用于parp-1的rnai序列:
[0055]5’‑
cgacctgatctggaacatcaa-3’(seq id no:1),以该rnai 序列为基础合成parp-1 shrna的上游序列片段parp1-f和下游序列片段 parp1-r。其中,所述parp1-f的核苷酸序列为:
[0056][0057]
所述parp1-r的核苷酸序列为:
[0058]058]
在seq id no:2和seq id no:3中的黑体部分对应的核苷酸序列为seq id no:1所示的目的序列或其反向重复序列,在seq id no:2和seq id no:3中带下划线的核苷酸序列可以形成loop环结构。seq id no:2和seq id no:3所示的序列在“退火”后会产生粘性末端,并能够和经过酶切后的载体连接。
[0059]
构建方法:(1)载体质粒plvx-shrna2-puro用ecor i bamh i双酶切,酶切反应在37℃水浴反应2h。
[0060]
(2)酶切后的质粒plvx-shrna2-puro回收大片段,并与parp1-shrna 的上游序列片段parp1-f和下游序列片段parp1-r的退火产物连接,22℃水浴反应过夜。
[0061]
连接体系如下:plvx-shrna2-puro大片段1μl,parp1-shrna退火产物1μl,10
×
dna ligase buffer 1μl(美国neb公司),t4 dna ligase 1μl(美国neb公司),ddh2o 6μl
[0062]
(3)取10μl过夜连接产物转化100μl jm109感受态细胞,将连接产物与感受态细胞混匀后冰浴30min,42℃热激45s,立即置冰上放置2min,加入预热至室温的400μl lb培养基,200rpm,37℃恒温摇床培养1h,4000rpm离心 1min,弃去400μl培养上清,剩余100μl用移液器混匀后均匀涂布于含100μg/ml 氨苄青霉素(ampicillin)抗性的lb平板上,倒置,37℃恒温培养箱培养过夜。
[0063]
(4)分别挑取单菌落接种于含5ml,100μg/ml ampicillin抗性的lb培养液中,250rpm,37℃恒温摇床培养过夜,用质粒抽提试剂盒抽提质粒,将重组质粒命名为plvx-shrna2-puro-parp-1-kd,并用xhoi做酶切鉴定。挑取酶切鉴定正确的阳性菌进行测序。
[0064]
试验结果:(1)将plvx-shrna2-puro-parp-1-kd重组质粒提取浓缩后,用xhoi做酶切鉴定,由于在载体骨架上含有一个xhoi酶切位点,结合插入片段上的xhoi位点可切出1320bp的条带,挑取3个克隆经鉴定后均为阳性克隆,鉴定酶切图谱如图1所示。
[0065]
(2)挑取酶切鉴定正确的一株阳性克隆进行核酸测序,测序结果显示插入片段的序列与所设计的parp-1 shrna的上游序列片段和下游序列片段的序列一致(具体测序结果参见图2)。
[0066]
综合酶切鉴定结果及核酸测序结果,证明载体质粒构建成功。
[0067]
实施例2慢病毒的包装
[0068]
传代hek293t细胞:按照6
×
106cells/培养皿的密度接种到10cm的细胞培养皿中,在37℃、5%co2培养箱中培养24h,用5ml不含青链霉素、含10%胎牛血清的dmem培养基在转染前2h换液。使用het高效转染试剂盒 bw11002(深圳百恩维生物科技有限公司)按照以下步骤完成慢病毒的包装:
[0069]
首先在a管中加入500μl het buffer a,在b管中加入10μl实施例1中制备的质粒plvx-shrna2-puro-parp-1-kd、15μl pspax2、pmd2.g包装质粒混合物(优宝生物)、50μl het buffer b、425μl ddh2o并混匀,将b管中的液体缓缓逐滴加入到a管中,用移液器轻轻
混匀并静置30min,然后将液体加入到培养皿中,轻轻倾斜混匀,于培养箱中培养16h后弃去培养基后换液为完全培养基dmem 10%fbs。
[0070]
继续培养过夜后收集上清保存于4℃冰箱,重新加入10ml完全培养基 dmem 10%fbs,过夜后再次收集上清,与前一天的上清混合后1000rpm离心5min,上清液以0.45μm pvdf滤器过滤至50ml圆底离心管中。4℃,20000g 离心4h,弃去上清后加入200μl/10cm培养皿的dmem溶液,重悬病毒沉淀,室温静置2h,然后用移液器轻轻地吹匀,继续室温放置30min,按每次使用的病毒量分装到洁净的1.5ml ep管中,-80℃冰箱保存待用。
[0071]
试验结果:将慢病毒载体质粒及pspax2、pmd2.g包装质粒混合物转染至hek293t细胞,换液前拍照。结果显示,gfp表达较高(具体参见图3),证明质粒表达成功。于换液后24h及48h收集病毒,浓缩后分装于-80℃冰箱待用。
[0072]
实施例3慢病毒的转染至hacat细胞moi值的确定
[0073]
moi(multiplicity of infection,感染复数)代表病毒与细胞数量的比值。
[0074]
主要步骤如下:在96孔培养板接种若干孔,每个孔内接种10000个目的细胞,按不同moi值(具体分别为1、10和100)加入病毒,二氧化碳培养箱 (37℃、5%co2)孵育24小时。孵育后吸去含病毒的培养液,用正常培养液(dmem 10%fbs)培养过夜。观察gfp荧光表达,并关注细胞生长状态,如有必要进行换液,以保证细胞良好的生长状态。
[0075]
根据细胞的生长状况、荧光表达情况综合判断病毒在哪个稀释度下作用效果最好(即最适的moi值),并以此稀释度下的病毒浓度作为后继实验的依据。
[0076]
根据细胞接种数量考察三种病毒量对hacat细胞的转染效率,对应的 moi值分别是100、10、1。有gfp表达的细胞代表病毒转染成功。
[0077]
结果显示,moi值在100时转染hacat细胞效率最高,约为10%;moi 为10时转染效率小于5%;moi为1时几乎没有gfp荧光表达。而过高的moi 值会造成明显的细胞毒性,且浪费病毒颗粒,因此确定后续转染时病毒与 hacat细胞的比率为100:1(具体参见图4)。
[0078]
实施例4嘌呤霉素浓度的确定
[0079]
24孔板内以5
×
104cells/孔的密度铺板。准备含不同浓度嘌呤霉素(0μg/ml, 1μg/ml,3μg/ml,6μg/ml,共4个梯度)的含10%fbs的dmem/f12培养基,细胞孵育过夜后加入上述培养基,孵育细胞,约2-3天更换新鲜的含不同浓度嘌呤霉素(0μg/ml,1μg/ml,3μg/ml,6μg/ml,共4个梯度)的含10%fbs 的dmem/f12培养基。每日监测细胞观察存活细胞比例,嘌呤霉素的最佳作用时间一般在1-4天之间,最小的抗生素使用浓度就是指:从抗生素筛选开始1-4天内杀死所有细胞的最低筛选浓度。
[0080]
在接种到24孔板的hacat细胞中分别加入0μg/ml,1μg/ml,3μg/ml, 6μg/ml 4个浓度的嘌呤霉素,观察48h后细胞死亡情况。结果发现,3μg/ml 以上的嘌呤霉素可使hacat全部死亡(具体参见图5),确定后续筛选单克隆细胞时嘌呤霉素的浓度为3μg/ml。
[0081]
实施例5稳定表达parp-1 shrna的单克隆hacat细胞系的筛选
[0082]
将实施例2中准备好的经过包装的病毒颗粒以100:1的数量比例加入到 hacat细胞中,24h后加入3μg/ml嘌呤霉素,筛选过程中可见到细胞大量死亡, 4-7d后适时转移到6孔板中,待细胞长满后至培养瓶中扩大培养,长满后即得到parp-1敲除/敲降的hacat细胞混合克隆细胞系。将细胞稀释到10个细胞每毫升,96孔板中每孔接种100μl,待细胞贴壁生长后,倒置显微镜下观察,标记只含有1个细胞的培养孔编号,记录细胞生长情况,15天左
右,即可挑选单克隆细胞株。
[0083]
在获得parp-1敲除/敲降的hacat细胞混合克隆细胞系后,通过极限稀释法,重新接种到96孔板中。理论上每孔只有一个细胞,在显微镜下标注只有一个细胞的孔,待15d后获得4株单克隆细胞株(具体参见图6)。
[0084]
实施例6parp-1表达量的蛋白质印迹法(western blot)验证
[0085]
采用含蛋白酶抑制剂的ripa细胞裂解液分别提取空白对照和实施例5中制备的parp-1敲除/敲降的hacat四个单克隆细胞系a-d的总蛋白,并用北京普利莱蛋白定量试剂盒(bca法)进行蛋白定量。
[0086]
其中,所述空白对照(简称为nc)为没有转染慢病毒的hacat细胞。提取所述单克隆细胞系中总蛋白的具体方法如下:
[0087]
(1)小心吸去培养液,用pbs冲洗细胞2次。最后一次彻底吸干残留液。
[0088]
(2)加入适当体积的ripa裂解液(含roche complete
tm
蛋白酶抑制剂(1 x))于培养皿、冰上孵育3-5分钟。期间反复晃动培养皿,使裂解液与细胞充分接触。
[0089]
(3)用细胞刮刀将细胞刮下,收集到1.5ml离心管中。
[0090]
(4)冰浴15min使细胞完全裂解。
[0091]
(5)12000g离心5min,收集上清,即为总蛋白溶液。
[0092]
根据蛋白定量结果,取25μg蛋白进行western blot(具体方法可参见《分子克隆指南》)。将所述蛋白样品上样sds-page凝胶,电泳分离后采用湿法电转到pvdf膜上,经过5%bsa封闭1h后,分别加入parp-1抗体(1:2000稀释)及β-actin抗体(1:10000稀释),4℃孵育过夜。加入hrp标记的山羊抗兔或山羊抗鼠二抗(1:10000稀释)室温孵育1h,ecl显色后于化学发光仪中曝光并记录结果。
[0093]
结果显示,与空白对照(即图7中所示的nc)组比较,a-d四株单克隆细胞系中的parp-1蛋白含量明显降低(具体参见图7),可用于后续研究。
[0094]
实施例7 parp-1敲降对dna损伤的影响
[0095]
将没有转染慢病毒的hacat细胞(简称为con)与实施例5中制备的稳定敲降parp-1的hacat单克隆细胞(简称为parp-1kd)分别接种于6孔板中,接种浓度为3
×
105个细胞/孔,采用10%胎牛血清 dmem/f12培养基培养。
[0096]
试验分组:未处理组(untreated)中不添加dna损伤剂硫芥(sulfur mustard, sm);试验组于细胞贴壁24h后加入dna损伤剂硫芥(sulfur mustard,sm), sm的添加量分别设定为10-4
m以及10-3
m,孵育6h及24h后两个时间点吸除培养液,用冷的pbs清洗一次,按照公司3-plex late apoptosis magnetic bead kit 96-well plate assay试剂盒说明书处理细胞并将样本冻存于-70℃冰箱,将两次样品统一上机检测hacat细胞中γ-h2ax的含量。
[0097]
结果显示:sm作用后,hacat细胞中γ-h2ax的含量明显增加,parp-1 敲降后会进一步增加hacat细胞中γ-h2ax的含量(具体参见图8)。γ-h2ax 作为dna损伤的指示蛋白,说明parp-1敲降显著性增加了dna损伤,提示 parp-1在正常皮肤细胞hacat的dna损伤修复中起到重要作用。
[0098]
讨论:
[0099]
本发明以慢病毒表达质粒plvx-shrna2-puro为载体,针对parp-1 mrna设计干扰
序列shrna,化学合成后采用酶切法插入上述慢病毒表达质粒中,测序及酶切结果显示包含parp-1 shrna的慢病毒载体质粒构建成功。
[0100]
将慢病毒载体质粒及pspax2、pmd2.g包装质粒混合物共转至hek293t 细胞,包装并提取用于后续实验的慢病毒颗粒。并在hacat细胞上考察了慢病毒的moi值和嘌呤霉素浓度,采用合适的慢病毒浓度转染至hacat细胞,筛选出4株单克隆细胞株,并使用western blot方法检测各株细胞的parp-1表达量,成功建立了稳定敲降parp-1蛋白的hacat单克隆细胞株。
[0101]
本发明采用的慢病毒载体转染hacat细胞,与现有技术中sirna介导的瞬时转染敲降基因的方式不同,本发明中parp-1的shrna序列可以通过慢病毒整合到细胞基因组中,得到稳定表达parp-1 shrna的细胞系。
[0102]
此外,多克隆细胞系构建成功后由于不同细胞克隆生长速度不一致, parp-1敲降效率高的细胞比敲降低的细胞生长缓慢,多次传代会导致 parp-1敲降效率高的细胞消失,parp-1敲降失败。本发明通过极限稀释法筛选得到parp-1敲降的单克隆细胞系,可以在多次传代中保持稳定,为后续研究提供可靠的细胞模型。
[0103]
目前,皮肤癌研究中常用的细胞模型多为癌细胞,本研究中选用的hacat细胞是人永生化表皮角化形成细胞,是皮肤相关研究中常用的正常细胞模型
【11、12
】。本方法建立的稳定敲降parp-1的hacat细胞模型为研究parp-1在正常皮肤dna损伤修复中的功能奠定基础;同时也能够为正常皮肤细胞dna损伤防治药物的筛选提供了有力支撑,筛选得到的化合物可作为皮肤癌预防及治疗的候选药物,为后续研究奠定基础。
[0104]
【参考文献】
[0105]
1.sharon,e.et al.laser treatment for non-melanoma skin cancer:a systematic review and meta-analysis.am j clin dermatol, doi:10.1007/s40257-020-00562-8(2020).
[0106]
2.shay,j.w.new insights into melanoma development.science(new york, n.y.)357,1358-1359,doi:10.1126/science.aao6963(2017).
[0107]
3.premi,s.et al.photochemistry.chemiexcitation of melanin derivatives induces dna photoproducts long after uv exposure.science(new york,n.y.) 347,842-847,doi:10.1126/science.1256022(2015).
[0108]
4.gracia-cazana,t.,gonzalez,s.,parrado,c.,juarranz,a.&gilaberte,y. influence of the exposome on skin cancer.actas dermosifiliogr 111,460-470, doi:10.1016/j.ad.2020.04.008(2020).
[0109]
5.yoon,j.h.et al.error-prone replication through uv lesions by dna polymerase theta protects against skin cancers.cell 176,1295-1309 e1215, doi:10.1016/j.cell.2019.01.023(2019).
[0110]
6.hegedus,c.et al.parp1 inhibition augments uvb-mediated mitochondrial changes-implications for uv-induced dna repair and photocarcinogenesis.cancers 12,doi:10.3390/cancers12010005(2019).
[0111]
7.daniels,c.m.,ong,s.e.&leung,a.k.the promise of proteomics for the study of adp-ribosylation.molecular cell 58,911-924, doi:10.1016/
j.molcel.2015.06.012(2015).
[0112]
8.alemasova,e.e.&lavrik,o.i.poly(adp-ribosyl)ation by parp1: reaction mechanism and regulatory proteins.nucleic acids research 47, 3811-3827,doi:10.1093/nar/gkz120(2019).
[0113]
9.mateo,j.et al.a decade of clinical development of parp inhibitors in perspective.annals of oncology:official journal of the european society for medical oncology 30,1437-1447,doi:10.1093/annonc/mdz192(2019).
[0114]
10.ma,w.,halweg,c.j.,menendez,d.&resnick,m.a.differential effects of poly(adp-ribose)polymerase inhibition on dna break repair in human cells are revealed with epstein-barr virus.proceedings of the national academy of sciences of the united states of america 109,6590-6595, doi:10.1073/pnas.1118078109(2012).
[0115]
11.wilson,v.g.growth and differentiation of hacat keratinocytes. methods in molecular biology(clifton,n.j.)1195,33-41, doi:10.1007/7651_2013_42(2014).
[0116]
12.yeo,h.,ahn,s.s.,lee,y.h.&shin,s.y.regulation of pro-opiomelanocortin(pomc)gene transcription by interleukin-31 via early growth response 1(egr-1)in hacat keratinocytes.molecular biology reports 47,5953-5962,doi:10.1007/s11033-020-05668-0(2020)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。