金属有机材料和制备方法
本技术是申请日为2014年7月13日的申请号为201480040099.6的中国专利申请的分案申请。
技术领域
1.本发明提供了包含多吡啶有机配体和在结构上与所述配体配位的金属离子的金属有机材料以及其制备方法。
2.缩写:afm,原子力显微镜;cp,配位聚合物;dmf,二甲基甲酰胺;eds,能量色散光谱法;fc,场冷却;ft-ir,红外傅里叶变换分光光度计;mof,金属有机框架;rt,室温;rtp,快速热处理;saed,选区电子衍射;sem,扫描电子显微镜;squid,超导量子干涉器件;tem,透射电子显微镜;tga,热重分析;xrd,x射线衍射;zfc,零场冷却。
背景技术:
3.有关金属有机框架(mof)以及配位聚合物(cp)的研究(对于全面了解术语cp、mof以及混合有机-无机材料之间的区别,参见biradha等,2009)已经由化学家和物理学家的科学团体以非常大的关注来对待,这是因为这些分子组装体的可调特性(通过控制它们的生长、尺寸以及形状)以及它们在催化、储气、分离、识别和纯化、光学器件、传感器等领域的潜在应用(zhao等,2004;yaghi等,2003;seo等,2000;kitagawa等,2004;evans和lin,2002;rowsell和yaghi,2005;tabellion等,2001;lei等,2007;zhao等,2008;chen等,2010)。在1964年,j.c.bailar定义了术语“配位聚合物”(bailar,1964)并且多种多样的技术,如溶剂热法(jung和oh,2008;ni和masel,2006)、沉淀法(oh和mirkin,2005;oh和mirkin,2006;sun等,2005;park等,2006;wei等,2007)以及反相微乳液法(rieter等,2006)已经被用于产生形状选择性纳米和微观结构化cp(wang等,2009;shi等,2011;liu等,2010;lu等,2011;li等,2011;cho等,2008)。
4.结构均匀度是用于常常在尺寸受限方案中涉及各种材料的定向制造的许多现实世界应用的一个先决条件(tuxen等,2013)。同时,结构多样性可以使得对所希望的物理特性和化学特性加以控制(noorduin等,2013;pevzner等,2012;whitesides和grzybowski,2002;masoomi和morsali,2013;gu等,2012)。分子自组装允许构建具有独特的结构和特性的复合上层结构。这样的复合材料的尺寸和形状受限合成因它们固有的和复杂的多功能性而是有利的,允许解决单个组分和其组合的特性以及它们空间整合到器件中以及表面上的可能性(carn
é‑sá
nchez等,2014)。当然,“在所有尺度下结构决定了功能”(tao等,2008)。
5.由于mof的独特的往往多孔的结构和经由合成可调性所实现的特殊的特性,所述mof已经在过去的几十年内被积极地研究(furukawa等,2013;cook等,2013;long和yaghi,2009)。然而,在微观和纳米水平上对它们的空间拓扑结构的控制仍是有限的并且难以实现的(stock和biswas,2011;sindoro等,2014)。许多变量,例如阴离子、溶剂、以及电子构型在形成几何上明确限定的和均匀的形状中起关键的作用。迄今,mof的形状限于简单的多面体(sindoro等,2014)。
技术实现要素:
6.根据本发明,已经发现以高均匀度具有不同的三维(亚微)微观结构的金属有机材料,特别是包含四面体多吡啶配体和与其配位的过渡金属离子的这样的材料可以通过特定的溶剂热合成来制备,同时对所述微观结构的均匀度和拓扑结构进行控制而不添加任何表面活性剂或调节剂。
7.在一个方面,本发明因此提供了一种金属有机材料,所述材料包含至少两个配体、在结构上与所述配体配位的至少两个金属离子、以及抗衡阴离子,其中所述配体中的每一个具有通式i:i r1(r
2-r
3-r4)4,其中r1是c,即或金刚烷-1,3,5,7-四基,即r2和r3各自独立地是不存在的,或选自(c
1-c8)亚烷基、(c
2-c8)亚烯基、(c
2-c8)亚炔基、亚环烷基、杂亚环烷基、亚芳-二基、杂亚芳-二基、或-n=n-,其中所述亚烷基、亚烯基、亚炔基、亚环烷基、杂亚环烷基、亚芳-二基以及杂亚芳-二基可以任选地被各自独立地选自以下各项的一个或多个基团取代:卤素、-or6、-cn、-cor6、-coor6、-con(r6)2、-ocoor6、-ocon(r6)2、-(c
1-c4)烷基、-o-(c
1-c4)烷基、-(c
1-c4)亚烷基-coor6、-n(r6)2、-no2、-sr6、-so2r6、或-s(=o)r6,或所述亚烷基、亚烯基以及亚炔基可以任选地被选自s、o或n的一个或多个相同的或不同的杂原子和/或选自以下各项的至少一个基团间断:-n=n-、-nh-co-、-co-nh-、-n(c
1-c4烷基)-、-n(c
6-c
10
芳基)-、或-(c
6-c
10
)亚芳-二基-,其中r6各自独立地是h、(c
1-c4)烷基、(c
2-c4)烯基或(c
2-c4)炔基;r4各自独立地是式ii的吡啶基、式iii的2,2'-联吡啶、或式iv的2,2':6',2
”‑
三联吡啶,它是经由其碳原子连接的;以及r5各自独立地是h、-cooh、-cn、-oh或-nh2。
8.在另一个方面,本发明涉及一种用于制备如上文所限定的金属有机材料的方法,所述方法包括以下步骤:(i)在压力容器,例如玻璃压力容器中提供(a)由阴离子和所述金属离子组成的金属盐的有机溶液或悬浮液;以及(b)所述配体的有机溶液或悬浮液;(ii)将所述压力容器密封并且在避光的情况下并且在没有搅拌的情况下将所述压力容器保持一段时间,任选地同时加热到60℃至120℃范围内的温度,持续整个所述时间段或其一部分,然后逐步冷却,从而使所述金属离子与所述配体反应以获得呈沉淀物形式的所述金属有机材料;以及(iii)收集所述沉淀物。
9.在又另一个方面,本发明涉及如上文所限定的金属有机材料用作用于气体吸附或气体分离的工艺中的吸附剂的用途。
10.在再另一个方面,本发明涉及一种用于气体吸附或气体分离的方法,所述方法是通过使所述气体吸附到吸附剂上来实现的,其中改进之处在于所述吸附剂是如上文所限定的金属有机材料。
附图说明
11.图1a-1b示出了(1a)砖状微观结构(nicll1)的sem图像;以及(1b)所述微砖的长度和宽度的分布。每一个条柱对应于在时间间隔xμm至(x 0.2)μm内的计数。afm测量表明200nm至300nm的厚度。
12.图2a-2b示出了砖状微观结构(nicll1)的tem图像(2a);以及示出了衍射光栅的tem图像(插图:电子衍射,比例尺=1nm-1
)(2b)。
13.图3a-3d示出了nicll1的afm表面特征(比例尺=1μm)(3a);单个雏晶的对应于3a中的垂直线的高度曲线(3b);nibrl2的afm表面特征(3c);以及单个雏晶的对应于3c中的水平线的高度曲线(3d)。
14.图4示出了如通过afm所测量的横跨随机选择的砖状微观结构中的一个的杨氏模量(young's modulus)。
15.图5示出了l1和nicll1的ft-ir谱(kbr压片)。
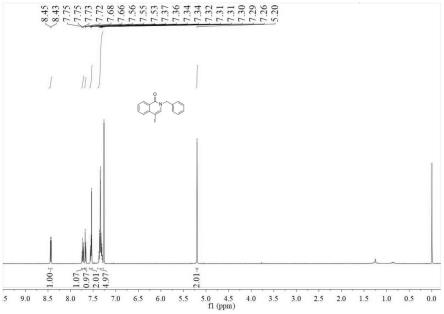
16.图6示出了在使nicll1与浓hcl反应并且用et3n中和之后chcl3萃取物的代表性1h nmr谱(cdcl3)。该谱对应于纯配体l1。
17.图7示出了在使用tem(120kv)的情况下nicll1的砖状微观结构的代表性eds测量结果。
18.图8示出了在以下时间立即对砖状结构nicll1的形成进行的与时间有关的sem分析(左图)以及描绘nicll1形成的草图(右图):在将nicl2·
6h2o的chcl3/dmf溶液与l1混合时(上图);在将反应混合物在密封的压力管中在105℃加热48小时后(中图);以及在将反应混合物加热5天并且受控冷却到室温之后(下图)(比例尺=2μm)。
19.图9示出了nicll1的代表性xrd谱。
20.图10示出了使用透镜内检测器(in lens detector)所拍摄的nicll1的sem图像。插图:nicll1的光学显微镜图像,所述图像确认了微观结构。
21.图11示出了sem图像,所述sem图像示出了在室温下在空气中在避光的情况下在以下各项中悬浮2个月的nicll1的砖状微观结构:dmf(图a);水(图b);或1:1v/v的dmf/水混合物(图c)。
22.图12示出了微观结构的sem图像,所述微观结构是通过在密封的压力管中在105℃在没有搅拌的情况下并且在避光的情况下将nicl2·
6h2o(1.6mg,6.9μmol)的dmf溶液(3.0ml)和l1(5.0mg,6.9μmol)的chcl3溶液(1.0ml)加热5天而获得的(图a);以及放大的图像(图b)。
23.图13示出了溶剂对nicll1的微观结构的影响。sem图像:dmf:chcl
3 3:1(v/v)(图a);dmf(图b);dmf:chcl
3 2:1(v/v)(图c);以及dmf:chcl
3 3:1(v/v) 0.5ml h2o(图d)(比例尺=10μm)。在溶剂热条件下在105℃使用l1(5.0mg,6.9μmol)的chcl3溶液和nicl2·
6h2o
(3.2mg,13.8μmol)的dmf溶液(总体积:4ml)将每一个实验进行5天。
24.图14示出了在10%h2/n2流下在300℃(图a)、400℃(图b)、500℃(图c)以及600℃(图d)对nicll1进行rtp 5分钟之后的sem图像。插图:ni纳米粒子的放大图像,比例尺=100nm。
25.图15示出了在10%h2/n2流下在200℃(图a)、300℃(图b)、400℃(图c)以及500℃(图d)在硅衬底上对nicll1进行rtp 5分钟之后的sem图像。
26.图16示出了在5分钟内对nicll1进行的rtp——在10%h2/n2流下和在真空中的稳定性的比较。sem图像:(图a)400℃、10%h2/n2;(图b)500℃、10%h2/n2;(图c)400℃、真空;(图d)500℃、真空。
27.图17示出了nicll1从30℃至1000℃的tga曲线(连续线)和温度曲线(虚线)。
28.图18示出了nicll1的甲烷吸附-解吸等温线。在室温、0℃以及-78.5℃暴露于甲烷之前,通过在120℃进行真空处理来活化样品。在室温下吸附等温线(实心红色圆圈)和解吸等温线(空心红色圆圈)。在0℃下吸附等温线(实心蓝色圆圈)和解吸等温线(空心蓝色圆圈)。在-78.5℃下吸附等温线(实心绿色圆圈)和解吸等温线(空心绿色圆圈)。
29.图19示出了在70℃、100℃以及120℃活化的nicll1对h2的吸附等温线(在78.5k)。在70℃活化之后的吸附等温线(实心红色圆圈)和解吸等温线(空心红色圆圈)。在100℃活化之后的吸附等温线(实心绿色圆圈)和解吸等温线(空心绿色圆圈)。在120℃活化之后的吸附等温线(实心蓝色圆圈)和解吸等温线(空心蓝色圆圈)。
30.图20a-20c示出了nicll1的摩尔磁化率的zfc和fc温度依赖性(20a);在t=6k下磁矩的磁场依赖性(20b);以及ni
2
相对于温度的计算的有效磁矩(20c)。
31.图21示出了nibrl2的磁性曲线(zfc和fc测量结果)。
32.图22a-22c示出了通过溶剂热合成所获得的含有氯化镍的微观结构的拓扑结构。22a-22b示出了nicll1的sem图像(尺寸:长度:2.6
±
0.9μm,宽度:1.4
±
0.5μm,厚度:250
±
50nm);并且22c图示了示出了nicll1的尺寸分布的直方图。反应条件:nicl2:l1=2:1,dmf/chcl3=3:1v/v,105℃,5天。
33.图23示出了通过溶剂热合成所获得的含有溴化镍的微观结构的拓扑结构。图a和b示出了nibrl2的sem图像。尺寸:对角线:370
±
10nm,边到边:405
±
10nm,厚度:220
±
20nm(图c)。反应条件:nibr2:l2=2:1,dmf/chcl3=3:1v/v,105℃,5天。
34.图24示出了前体的摩尔比(图a-b)和溶剂(图c-f)的变化对nicll1的结构的影响。图a和b分别示出了使用1:1的nicl2与l1的比率所获得的微观结构的sem图像和其放大图像;反应条件:nicl2·
6h2o(6.9μmol)的dmf溶液(3.0ml)、l1(6.9μmol)的chcl3溶液(1.0ml),105℃,5天,在没有搅拌的情况下以及在避光的情况下。图c、d、e以及f分别示出了使用以下各项所获得的nicll1的sem图像:dmf:chcl
3 3:1(v/v);dmf;dmf:chcl
3 2:1(v/v);以及dmf:chcl
3 3:1(v/v) 0.5ml h2o;反应条件:nicl2·
6h2o(13.8μmol)的dmf溶液、l1(6.9μmol)的chcl3溶液,105℃,5天,在没有搅拌的情况下以及在避光的情况下。
35.图25示出了含镍的微观结构的tem图像和saed。图a:nicll1的tem图像;图b:示出了nicll1的单晶中的晶格面的高倍放大tem图像,插图:源自于nicll1的saed图,比例尺=2nm-1
,其中d间距对应于1:1.79nm、2:0.9nm、3:0.46nm、4:0.42nm、5:0.49nm;图c:nibrl2的tem图像;以及图d:源自于nibrl2的saed图,其中d间距对应于1':0.95nm、2':0.49nm、3':
0.55nm。对于saed,晶体取向(最长的轴)在图a和c中由黄色箭头所指示。
36.图26示出了在使用tem(120kv)的情况下nibrl2的代表性eds。
37.图27示出了在使cubrl2与浓hcl反应并且用et3n中和之后chcl3萃取物的代表性1h nmr谱(cdcl3)。该谱对应于纯l2。
38.图28示出了nicll1的rtp。图a-e:在10%h2/n2下进行rtp(插图比例尺=100nm)。图f-g:在真空下进行rtp。对被滴铸在硅衬底中的nicll1进行实验,持续5分钟。
39.图29示出了nibrl2的rtp。图a-c:在10%h2/n2下进行rtp(插图比例尺=60nm)。图d-f:在真空下进行rtp。对被滴铸在硅衬底中的nibrl2进行实验,持续5分钟。
40.图30示出了cucll2的sem图像。反应条件:1当量l2、2当量cucl2、3:1(v/v)dmf/chcl3,105℃,5天。
41.图31示出了通过溶剂热合成所获得的含有溴化铜的mof的互穿拓扑结构。cubrl2的sem图像(图a-c)。反应条件:cubr2:l2=2:1,dmf/chcl3=3:1v/v,105℃,5天。
42.图32示出了通过溶剂热合成所获得的含有硝酸铜的mof的互穿拓扑结构。cu(no3)l2的代表性sem图像(图a-c)。反应条件:cu(no3)2:l2=1:1,dmf/chcl3=3:1v/v,105℃,5天。
43.图33示出了cu(no3)l2的sem图像的实例(1当量l2、1当量cu(no3)2、3:1(v/v)dmf/chcl3,105℃,5天)。
44.图34a-34b示出了在惰性气氛下通过溶剂热合成所获得的含有硝酸铜的mof的矩形拓扑结构。(34a)cu(no3)l2的代表性sem图像。(34b)示出了cu(no3)l2的尺寸分布的直方图。长度:3.65
±
0.95μm,宽度:0.675
±
0.09μm。反应条件(使用无水溶剂,在氮气下):cu(no3)2:l2=1:1,dmf/chcl3=3:1v/v,105℃,5天。
45.图35示出了l2、cu(no3)l2以及cubrl2(kbr压片)的ft-ir谱。
46.图36示出了溶剂和温度的变化对cu(no3)l2的结构的影响。使用dmso/chcl3(4ml,3:1v/v)在60℃所获得的结构(图a);以及使用phcn/chcl3(4ml,3:1v/v)在60℃所获得的结构(图b)的sem图像。反应条件:cu(no3)2·
3h2o(3.3mg,13.6μmol)、l2(5.0mg,6.8μmol)的chcl3溶液(1.0ml),60℃,5天,在没有搅拌的情况下以及在避光的情况下。在105℃获得类似的结构。
47.图37示出了溶剂和温度的变化对cubrl2的结构的影响。使用以下各项所获得的结构的sem图像:phcn/chcl3(4ml,3:1v/v)在60℃下(图a);dmf/mecn/chcl3(4ml,1.5:1.5:1v/v/v)在60℃下(图b);mecn/chcl3(4ml,3:1v/v)在60℃下(图c);以及dmso/chcl3(4ml,3:1v/v)在60℃下(图d)。反应条件:cubr(3mg,13.6μmol)、l2(5.0mg,6.8μmol)的chcl3溶液(1.0ml),60℃,5天,在没有搅拌的情况下以及在避光的情况下。在105℃获得类似的结构。
48.图38示出了对于基于ni的mof的形成进行的与时间有关的sem分析。nicll1:在将nicl2的dmf溶液和l1的chcl3溶液在室温混合后(图a);将这一混合物在105℃加热1天和5天后(分别是图b和c)立即分析。nibrl2:在将nibr2的dmf溶液和l2的chcl3溶液在室温混合后(图d);将这一混合物在105℃加热1天和5天后(分别是图e和f)立即分析。比例尺:(图a-c=2μm;图d-f=500nm;图d插图=200nm)。
49.图39示出了对于cubrl2的形成进行的与时间有关的sem分析。在以下各项之后获得的cubrl2的拓扑结构(图a-e;比例尺=5μm):(图a)在将cubr2的dmf溶液和l2的chcl3溶液
在室温混合后立即。插图比例尺=200nm;(图b)将这一混合物在105℃加热1.5天后立即。插图比例尺=2μm;(图c)将混合物在105℃加热2.5天后立即。插图比例尺=1μm;(图d、d')将混合物在105℃加热3.5天后立即。插图比例尺=2μm;以及(图e)将混合物在105℃加热5天后立即。插图比例尺=2μm。
50.图40示出了对于cu(no3)l2的形成进行的与时间有关的sem分析。在以下各项之后获得的cu(no3)l2的拓扑结构(图a-d;比例尺=5μm):(图a)将cu(no3)2的dmf溶液和l2的chcl3溶液在105℃加热1天。插图:在将cu(no3)2和l2混合之后立即。插图比例尺=200nm;(图b)将混合物在105℃加热2.5天;(图c)将混合物在105℃加热3.5天;以及(图d)将混合物在105℃加热5天。
51.图41示出了在研究3中所述的条件下由l1的chcl3溶液和nibr2的dmf悬浮液制备的mof。
52.图42示出了在研究3中所述的条件下由l1的chcl3溶液和nicl2·
6h2o的dmf悬浮液制备的mof。
53.图43示出了在研究3中所述的条件下由l2的chcl3溶液和nicl2·
6h2o的dmf溶液制备的mof的sem图像和tem图像(分别是图a和b)。
54.图44示出了在研究3中所述的条件下由pd(cod)cl2的超声处理的甲苯溶液和l2的甲苯悬浮液制备的mof。
55.图45示出了在研究3中所述的条件下由pdcl2的超声处理的甲苯悬浮液和l2的甲苯悬浮液制备的mof。
56.图46示出了在研究3中所述的条件下由pd(phcn)2cl2的乙苯溶液和l2的乙苯悬浮液制备的mof。
57.图47示出了在研究3中所述的条件下由pd(phcn)2cl2的甲苯溶液和l2的庚烷悬浮液制备的mof。
58.图48示出了在研究3中所述的条件下由l2的chcl3溶液和cu(otf)2的dmf溶液制备的mof。
59.图49示出了在研究3中所述的条件下由l2的chcl3溶液和cu(otf)2的dmf溶液制备的mof。
60.图50示出了在研究3中所述的条件下由l2的无水chcl3溶液和cu(no3)2·
3h2o的无水dmf溶液制备的mof。
61.图51示出了在研究3中所述的条件下由l2的chcl3溶液和zn(oac)2·
2h2o的dmf溶液制备的mof。
62.图52示出了在研究3中所述的条件下由l2的chcl3溶液和zn(oac)2·
2h2o的dmf溶液制备的mof。
63.图53示出了在研究3中所述的条件下由l2的chcl3溶液和zncl2·
2h2o的dmf溶液制备的mof。
64.图54示出了在研究3中所述的条件下由l2的chcl3溶液和zncl2·
2h2o的dmf溶液制备的mof。
65.图55示出了在研究3中所述的条件下由l2的chcl3溶液和zncl2·
2h2o的dmf溶液制备的mof。
66.图56示出了在研究3中所述的条件下由l2的chcl3溶液和znbr2的dmf溶液制备的mof。
具体实施方式
67.在一个方面,本发明提供了如上文所限定的金属有机材料,即包含各自具有如上文所限定的通式i的至少两个配体、在结构上与所述配体配位的至少两个金属离子、以及抗衡阴离子的金属有机材料。
68.如本文所用的术语“金属有机材料”或“金属有机框架(mof)”指的是一种特定类型的配位聚合物,更特别是有机金属聚合物,它含有金属阳离子,优选地过渡金属阳离子,所述金属阳离子与各自具有通式i的有机配体配位以形成可以是多孔的一维结构、二维结构或三维结构,其中金属阳离子和有机配体的选择决定了结构以及因此决定了mof的特性。更具体地说,mof是具有含有潜在空隙的有机配体的配位网络,其中术语“配位网络”指的是经由使配位实体在一维上,但是通过两个或更多个单独的链、环之间的交联或螺环连接,或在二维或三维上重复所引起的配位低聚物延伸(还参见biradha等,2009)。
69.如本文所用的术语“卤素”包括氟、氯、溴以及碘。
70.如本文所用的术语“烷烃”指的是优选地具有5-14个碳原子的直链或支链、或环状(包括双环)饱和烃,并且包括例如戊烷、己烷、环己烷、庚烷、环庚烷、辛烷、环辛烷、壬烷、癸烷、十氢化萘等。
71.如本文所用的术语“烷醇”指的是优选地具有1-10个碳原子并且含有羟基/醇官能团(-oh)代替氢原子的烷烃,并且包括例如甲醇、乙醇、异丙醇、正丁醇、仲丁醇、异丁醇、戊醇、己醇等。
72.如本文所用的术语“烷基”通常意指优选地具有1-8个,更优选地具有1-4个碳原子的直链或支链烃基,并且包括例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、2,2-二甲丙基、正己基、正庚基、正辛基等。
73.如本文所用的术语“亚烷基”指的是优选地具有1-8个碳原子的直链二价烃链并且包括例如亚甲基、亚乙基、亚丙基、亚丁基、亚戊基、亚己基、亚庚基、亚辛基等。术语“亚烯基”和“亚炔基”通常分别意指优选地具有2-8个碳原子和至少一个双键或三键的直链二价烃基。这些亚烯基的非限制性实例包括亚乙烯基、1,3-亚丙烯基、1,4-亚丁-1-烯基、1,4-亚丁-2-烯基、1,4-亚丁-3-烯基、1,5-亚戊-1-烯基、1,5-亚戊-2-烯基、1,5-亚戊-3-烯基、1,5-亚戊-4-烯基、1,6-亚己-1-烯基、1,6-亚己-2-烯基、1,6-亚己-3-烯基、1,6-亚己-4-烯基、1,6-亚己-5-烯基、1,7-亚庚-1-烯基、1,7-亚庚-2-烯基、1,7-亚庚-3-烯基、1,7-亚庚-4-烯基、1,7-亚庚-5-烯基、1,7-亚庚-6-烯基、1,8-亚辛-1-烯基、1,8-亚辛-2-烯基、1,8-亚辛-2-烯基、1,8-亚辛-3-烯基、1,8-亚辛-4-烯基、1,8-亚辛-5-烯基、1,8-亚辛-6-烯基、1,8-亚辛-7-烯基等;并且这些亚炔基的实例包括而不限于亚乙炔基、1,3-亚丙炔基、1,4-亚丁-1-炔基、1,4-亚丁-2-炔基、1,4-亚丁-3-炔基、1,5-亚戊-1-炔基、1,5-亚戊-2-炔基、1,5-亚戊-3-炔基、1,5-亚戊-4-炔基、1,6-亚己-1-炔基、1,6-亚己-2-炔基、1,6-亚己-3-炔基、1,6-亚己-4-炔基、1,6-亚己-5-炔基、1,7-亚庚-1-炔基、1,7-亚庚-2-炔基、1,7-亚庚-3-炔基、1,7-亚庚-4-炔基、1,7-亚庚-5-炔基、1,7-亚庚-6-炔基、1,8-亚辛-1-炔基、1,8-亚辛-2-炔基、1,8-亚辛-2-炔基、1,8-亚辛-3-炔基、1,8-亚辛-4-炔基、1,8-亚辛-5-炔基、1,
8-亚辛-6-炔基、1,8-亚辛-7-炔基等。
74.如本文所用的术语“亚环烷基”通常意指优选地具有3-10个碳原子的单环或双环饱和二价烃基,如亚环丙基、亚环丁基、亚环戊基、亚环己基、亚环庚基、亚环辛基、亚环癸基、双环[3.2.1]辛烷-二基、双环[2.2.1]庚烷-二基等。术语“杂亚环烷基”指的是其中环碳原子中的至少一个被选自n、o或s的杂原子置换的亚环烷基。
[0075]
如本文所用的术语“芳基”表示优选地具有6-14个碳原子的由单个环或稠合的或通过共价键连接的多个环组成的芳族碳环基团,诸如但不限于苯基、萘基、菲基、以及联苯。术语“亚芳-二基”指的是通过从两个环碳原子去除氢原子而衍生自芳烃的二价基团。亚芳-二基的非限制性实例包括苯-1,3-二基、苯-1,4-二基、萘-二基、菲-2,7-二基、联苯-4,4'-二基等。
[0076]
术语“杂亚芳-二基”指的是通过从两个环原子去除氢原子而衍生自单环或多环杂芳环的二价基团,所述杂芳环含有一个至三个,优选地1-2个选自由n、o以及s组成的组的杂原子。当杂亚芳-二基是单环杂芳环时,它优选地是5元-6元环的二价基团,诸如但不限于吡咯-2,5-二基、吡咯-3,5-二基、呋喃-2,5-二基、呋喃-3,5-二基、噻吩-2,5-二基、噻吩-3,5-二基、噻嗪-2,5-二基、噻嗪-3,6-二基、吡唑-1,3-二基、吡唑-1,4-二基、吡唑-3,5-二基、吡嗪-2,5-二基、吡嗪-2,6-二基、咪唑-1,4-二基、咪唑-2,4-二基、咪唑-2,5-二基、噁唑-2,4-二基、噁唑-2,5-二基、异噁唑-3,5-二基、噻唑-2,4-二基、噻唑-2,5-二基、异噻唑-3,5-二基、吡啶-2,4-二基、吡啶-3,6-二基、嘧啶-2,4-二基、嘧啶-2,5-二基、1,2,3-三嗪-4,6-二基、1,3,4-三嗪-2,5-二基、1,3,4-三嗪-2,6-二基、1,3,5-三嗪-2,4-二基等。由两个环构成的多环杂亚芳-二基的实例包括而不限于苯并呋喃-2,5-二基、苯并呋喃-2,6-二基、异苯并呋喃-2,4-二基、异苯并呋喃-2,5-二基、苯并噻吩-2,5-二基、苯并噻吩-2,6-二基、吲哚-2,5-二基、吲哚-2,6-二基、喹啉-2,6-二基、喹啉-2,7-二基、喹啉-3,6-二基、喹啉-3,7-二基、异喹啉-3,6-二基、异喹啉-3,7-二基、咪唑并[1,2-a]吡啶-2,6-二基、咪唑并[1,2-a]吡啶-2,7-二基、苯并咪唑-2,5-二基、苯并咪唑-2,6-二基、苯并噻唑-2,5-二基、苯并噻唑-2,6-二基、苯并噁唑-2,5-二基、苯并噁唑-2,6-二基、吡啶并[1,2-a]嘧啶-2,7-二基、吡啶并[1,2-a]嘧啶-2,8-二基、吡啶并[1,2-a]嘧啶-3,7-二基、吡啶并[1,2-a]嘧啶-3,7-二基、1,3-苯并二噁英-2,6-二基、1,3-苯并二噁英-2,7-二基等。
[0077]
亚烷基、亚烯基、亚炔基、亚环烷基、杂亚环烷基、亚芳-二基以及杂亚芳-二基可以任选地被各自独立地选自以下各项的一个或多个基团取代:卤素、-or6、-cn、-cor6、-coor6、-con(r6)2、-ocoor6、-ocon(r6)2、-(c
1-c4)烷基、-o-(c
1-c4)烷基、-(c
1-c4)亚烷基-coor6、-n(r6)2、-no2、-sr6、-so2r6或-s(=o)r6,或所述亚烷基、亚烯基以及亚炔基可以任选地被一个或多个,例如一个或两个选自s、o或n的相同的或不同的杂原子和/或各自独立地选自以下各项的至少一个基团,例如一个、两个或三个基团间断:-n=n-、-nh-co-、-co-nh-、-n(c
1-c4烷基)-、-n(c
6-c
10
芳基)-或-(c
6-c
10
)亚芳-二基-,其中r6各自独立地是h、(c
1-c4)烷基、(c
2-c4)烯基或(c
2-c4)炔基。
[0078]
在某些实施方案中,本发明的金属有机材料是如上文所限定的通式i的材料,其中r2和r3各自独立地是不存在的,或选自(c
1-c8)亚烷基、(c
2-c8)亚烯基、(c
2-c8)亚炔基、亚环烷基、杂亚环烷基、亚芳-二基、杂亚芳-二基、或-n=n-,其中所述亚烷基、亚烯基、亚炔基、亚环烷基、杂亚环烷基、亚芳-二基以及杂亚芳-二基可以任选地被各自独立地选自以下各
项的一个或多个基团取代:卤素、-or6、-cn、-cor6、-coor6、-con(r6)2、-ocoor6、-ocon(r6)2、-(c
1-c4)烷基、-o-(c
1-c4)烷基、-(c
1-c4)亚烷基-coor6、-n(r6)2、-no2、-sr6、-so2r6、或-s(=o)r6,其中r6是h,或所述亚烷基、亚烯基以及亚炔基可以任选地被选自s、o或n的一个或多个相同的或不同的杂原子和/或选自以下各项的至少一个基团间断:-n=n-、-nh-co-、-co-nh-、-n(c
1-c4烷基)-、-n(c
6-c
10
芳基)-、或-(c
6-c
10
)亚芳-二基-。
[0079]
在某些具体的这样的实施方案中,r2和r3各自独立地是不存在的,或选自(c
1-c4)亚烷基、(c
2-c4)亚烯基、(c
2-c4)亚炔基、亚环烷基、杂亚环烷基、亚芳-二基、杂亚芳-二基、或-n=n-,其中所述亚烷基、亚烯基、亚炔基、亚环烷基、杂亚环烷基、亚芳-二基以及杂亚芳-二基可以任选地被以下各项取代:卤素、-oh、-cn、-coh、-cooh、-conh2、-ocooh、-oconh2、-(c
1-c2)烷基、-o-(c
1-c2)烷基、-(c
1-c2)亚烷基-cooh、-nh2、-no2、-sh、-so2h、或-s(=o)h,或所述亚烷基、亚烯基以及亚炔基可以任选地被选自s、o或n的一个或多个相同的或不同的杂原子和/或选自以下各项的至少一个基团间断:-n=n-、-nh-co-、-co-nh-、-n(c
1-c2烷基)-、-n(c6芳基)-、或-(c6)亚芳-二基-。
[0080]
在更具体的这样的实施方案中,本发明的金属有机材料是通式i的材料,其中r2和r3各自独立地是不存在的,或选自(c
1-c4)亚烷基、(c
2-c4)亚烯基、(c
2-c4)亚炔基、亚芳-二基、或杂亚芳-二基,例如其中(i)r2和r3中的一个是不存在的并且r2和r3中的另一个是(c
2-c4)亚烷基、(c
2-c4)亚烯基、(c
2-c4)亚炔基、亚芳-二基、或杂亚芳-二基;(ii)r2和r3中的一个是(c
2-c4)亚烷基、(c
2-c4)亚烯基或(c
2-c4)亚炔基,并且r2和r3中的另一个是亚芳-二基或杂亚芳-二基;或(iii)r2和r3这两者均是不存在的。某些具体的这样的实施方案是如下的那些实施方案,其中r2是(c
2-c4)亚烯基或(c
2-c4)亚炔基,并且r3是(c6)亚芳-二基;或r2是(c6)亚芳-二基,并且r3是(c
2-c4)亚烯基或(c
2-c4)亚炔基。
[0081]
在某些实施方案中,本发明的金属有机材料是如上文所限定的通式i的材料,其中r4各自独立地是式ii的吡啶基,其中r5各自独立地是h、-cooh、-cn、-oh或-nh2,优选地h或-cooh。
[0082]
在某些实施方案中,本发明的金属有机材料是如上文所限定的通式i的材料,其中r1是c或金刚烷-1,3,5,7-四基;r2和r3各自独立地是不存在的,或选自(c
1-c4)亚烷基、(c
2-c4)亚烯基、(c
2-c4)亚炔基、亚环烷基、杂亚环烷基、亚芳-二基、杂亚芳-二基或-n=n-,其中所述亚烷基、亚烯基、亚炔基、亚环烷基、杂亚环烷基、亚芳-二基以及杂亚芳-二基可以任选地被各自独立地选自以下各项的一个或多个基团取代:卤素、-or6、-cn、-cor6、-coor6、-con(r6)2、-ocoor6、-ocon(r6)2、-(c
1-c4)烷基、-o-(c
1-c4)烷基、-(c
1-c4)亚烷基-coor6、-n(r6)2、-no2、-sr6、-so2r6、或-s(=o)r6,其中r6是h,或所述亚烷基、亚烯基以及亚炔基可以任选地被选自s、o或n的一个或多个相同的或不同的杂原子和/或选自以下各项的至少一个基团间断:-n=n-、-nh-co-、-co-nh-、-n(c
1-c2烷基)-、-n(c6芳基)-或-(c6)亚芳-二基-;r4各自独立地是式ii的吡啶基;并且r5各自独立地是h、-cooh、-cn、-oh或-nh2,优选地h或-cooh。
[0083]
在某些具体的这样的实施方案中,r2和r3各自独立地是不存在的,或选自(c
1-c4)亚烷基、(c
2-c4)亚烯基、(c
2-c4)亚炔基、亚芳-二基或杂亚芳-二基;r4是经由氮原子对位的碳原子连接的式ii的吡啶基;并且r5是h或-cooh。在更具体的这样的实施方案中,本发明的金属有机材料是通式i的材料,其中(i)r2和r3中的一个是不存在的并且r2和r3中的另一个是(c
2-c4)亚烷基、(c
2-c4)亚烯基、(c
2-c4)亚炔基、亚芳-二基、或杂亚芳-二基;(ii)r2和r3中
的一个是(c
2-c4)亚烷基、(c
2-c4)亚烯基或(c
2-c4)亚炔基,并且r2和r3中的另一个是亚芳-二基或杂亚芳-二基;或(iii)r2和r3这两者均是不存在的,例如其中r2是(c
2-c4)亚烯基或(c
2-c4)亚炔基,并且r3是(c6)亚芳-二基;或r2是(c6)亚芳-二基,并且r3是(c
2-c4)亚烯基或(c
2-c4)亚炔基。某些具体的这样的实施方案是如下的那些实施方案,其中r2是(c6)亚芳-二基;并且r3是(c2)亚烯基或(c2)亚炔基,即如下的金属有机材料,所述材料包含在结构上与各自具有通式i的至少两个多吡啶配体配位的至少两个金属离子,所述配体由与四个相同的“臂”连接的c或金刚烷-1,3,5,7-四基组成,这四个相同的“臂”各自分别是(4-(2-(吡啶-4-基)乙烯基)苯基)或(4-(吡啶-4-基乙炔基)苯基)。
[0084]
在具体的实施方案中,本发明的金属有机材料是通式i的材料,其中(i)r1是c,并且所述配体中的每一个是四(4-(吡啶-4-基乙炔基)苯基)甲烷或四(4-(2-(吡啶-4-基)乙烯基)苯基)甲烷,它们在本文分别被标示为配体l1和l2;或(ii)r1是金刚烷-1,3,5,7-四基,并且所述配体中的每一个是1,3,5,7-四(4-(吡啶-4-基乙炔基)苯基)金刚烷或1,3,5,7-四(4-(2-(吡啶-4-基)乙烯基)苯基)金刚烷,它们在本文分别被标示为配体l3和l4(参见附录)。
[0085]
在某些实施方案中,本发明的金属有机材料内所包含的金属离子是过渡金属的离子,所述过渡金属诸如os、ru、fe、pt、pd、ni、ir、rh、zn、co、cu、re、tc、mn、v、nb、ta、hf、zr、cr、mo、w、ti、sc、ag、au、y或其组合。在具体的这样的实施方案中,所述金属离子是ni、cu、pd或zn中的一种或多种(即组合)的离子。
[0086]
在某些实施方案中,本发明的金属有机材料内所包含的抗衡阴离子选自无机阴离子、有机阴离子、或其组合。无机阴离子的实例包括而不限于f-、cl-、br-、i-、no
3-、pf
6-、bf
4-、oh-、clo
4-、so
3-、以及cn-;并且有机阴离子的非限制性实例包括烷基coo-,优选地乙酰氧基(oac)、cf3coo-、芳基coo-、三氟甲烷磺酸根(三氟甲磺酸根,otf)。
[0087]
如上文所限定,本发明的金属有机材料是一种特定类型的配位聚合物,所述配位聚合物含有金属离子,优选地过渡金属离子,所述金属离子与各自具有通式i的有机配体配位以形成一维结构、二维结构或三维结构。在某些实施方案中,本发明的金属有机材料包含在结构上配位于所述至少两个配体中的两个之间的所述金属离子中的至少一个。
[0088]
在某些实施方案中,本发明的金属有机材料包含金属离子,所述金属离子与各自具有通式i的有机配体配位以形成三维(3d)结构。在具体的这样的实施方案中,本发明的金属有机材料具有三维结晶微观结构或亚微结构,更特别是其中所述结晶微观结构或亚微结构具有例如砖状微观结构的几何形状。三维结晶几何形状的实例包括而不限于六边形、球形、星型八面体、以及花状形状。
[0089]
在总体上具有三维结构的mof,以及特别是根据本发明的这些mof是多孔的,并且因此还可能包含溶剂分子,所述溶剂分子也被称为“客体分子”,从制备过程中被留下并且被限制在三维结构的孔隙内。由于在消除那些溶剂分子期间mof的孔隙是稳定的,因此这些mof可以在用于气体吸附的工艺中用作吸附剂,例如h2、co、co2或甲烷吸附、或气体分离和/或纯化,例如将co2和甲烷分离。
[0090]
在一个具体的实施方案中,本发明的金属有机材料包含各自是配体l1的配体、在结构上与所述配体的氮原子配位的ni(ii)离子、以及作为抗衡阴离子的cl-,其中所述金属有机材料具有化学式(nicl2n2c
26.5h16
)n,其中n是至少4的整数,并且三维结晶微观结构或亚
微结构任选地还包含溶剂分子。这种材料在本文被标示为nicll1并且可以例如通过以下步骤而形成:在烘干的玻璃压力管中将l1的氯仿溶液添加到nicl2·
6h2o的二甲基甲酰胺(dmf)悬浮液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热5天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡绿色沉淀物形式的nicll1,所述nicll1具有细长的六边形(砖状)的几何形状。所获得的nicll1结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0091]
在另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l1的配体、在结构上与所述配体的氮原子配位的ni(ii)离子、以及作为抗衡阴离子的br-,其中所述金属有机材料具有化学式(nibr2n2c
26.5h16
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为nibrl1并且可以例如通过以下步骤而形成:在烘干的玻璃压力管中将l1的氯仿溶液添加到nibr2的dmf悬浮液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热5天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡绿色沉淀物形式的nibrl1。所获得的nibrl1结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0092]
在再另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的ni(ii)离子、以及作为抗衡阴离子的cl-,其中所述金属有机材料具有化学式(nicl2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为nicll2并且可以例如通过以下步骤形成:在烘干的玻璃管中小心地使l2的氯仿溶液在nicl2·
6h2o的dmf溶液下方分层,然后将所述玻璃管密封并且在暗处保持5天,从而获得呈淡绿色沉淀物形式的nicll2。所获得的nicll2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0093]
在又另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的ni(ii)离子、以及作为抗衡阴离子的br-,其中所述金属有机材料具有化学式(nibr2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为nibrl2并且可以例如通过以下步骤而形成:在玻璃压力管中将l2的氯仿溶液添加到nibr2的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热5天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡绿色沉淀物形式的nibrl2,所述nibrl2具有接近正六边形的几何形状。所获得的nibrl2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0094]
在另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的cu(ii)离子、以及作为抗衡阴离子的cl-,其中所述金属有机材料具有化学式(cucl2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为cucll2并且可以例如通过以下步骤而形成:在玻璃压力管中将l2的氯仿溶液添加到cucl2的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热5天;然后在9小时至10小时内逐步冷却到室温,从而获得呈深绿色沉淀物形式的cucll2。所获得的cucll2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0095]
在再另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配
体、在结构上与所述配体的氮原子配位的cu(ii)离子、以及作为抗衡阴离子的br-,其中所述金属有机材料具有化学式(cubr2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为cubrl2并且可以例如通过以下步骤而形成:在玻璃压力管中将l2的氯仿溶液添加到cubr2的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热5天;然后在9小时至10小时内逐步冷却到室温,从而获得呈深绿色沉淀物形式的cubrl2,所述cubrl2具有星型八面体的几何形状。所获得的cubrl2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0096]
在又另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的cu(ii)离子、以及作为抗衡阴离子的no
3-,其中所述金属有机材料具有化学式(cu(no3)2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为cu(no3)2l2并且可以例如通过以下步骤而形成:在玻璃压力管中将l2的氯仿溶液添加到cu(no3)2的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热5天;然后在9小时至10小时内逐步冷却到室温,从而获得呈深绿色沉淀物形式的cu(no3)2l2。或者,cu(no3)2l2可以通过以下步骤而形成:在烘干的玻璃压力管中在n2气氛下将l2的无水氯仿溶液添加到cu(no3)2·
3h2o的无水dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热6天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡绿色沉淀物形式的cu(no3)2l2,所述cu(no3)2l2具有矩形棱柱的几何形状。所获得的cu(no3)2l2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0097]
在另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的cu(ii)离子、以及作为抗衡阴离子的otf-,其中所述金属有机材料具有化学式(cu(otf)2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为cu(otf)2l2并且可以例如通过以下步骤形成:在烘干的玻璃管中小心地使l2的氯仿溶液在cu(otf)2的dmf溶液下方分层,然后将所述玻璃管密封并且在暗处保持5天,从而获得呈淡蓝色沉淀物形式的cu(otf)2l2。或者,cu(otf)2l2可以通过以下步骤而形成:在烘干的玻璃管中小心地使l2的氯仿溶液在cu(otf)2的dmf溶液下方分层,然后将所述玻璃管密封并且在暗处保持10天;在60℃在没有搅拌的情况下并且在避光的情况下再加热2天;然后逐步冷却到室温,从而获得呈淡蓝色沉淀物形式的cu(otf)2l2。所获得的cu(otf)2l2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0098]
在再另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的pd(ii)离子、以及作为抗衡阴离子的cl-,其中所述金属有机材料具有化学式(pdcl2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为pdcll2并且可以例如通过以下步骤而形成:在烘干的玻璃压力管中将pd(cod)cl2的经过超声处理的甲苯溶液添加到l2的甲苯悬浮液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈黄色沉淀物形式的pdcll2。在一种替代性方法中,pdcll2可以通过以下步骤而形成:在烘干的玻璃压力管中
将pdcl2的经过超声处理的甲苯悬浮液添加到l2的甲苯悬浮液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈黄色沉淀物形式的pdcll2。在另一种替代性方法中,pdcll2可以通过以下步骤而形成:在烘干的玻璃压力管中将pd(phcn)2cl2的乙苯溶液添加到l2的乙苯悬浮液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈黄色沉淀物形式的pdcll2。在另一种替代性方法中,pdcll2可以通过以下步骤而形成:在烘干的玻璃压力管中将pd(phcn)2cl2的甲苯溶液添加到l2的庚烷悬浮液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈黄色沉淀物形式的pdcll2。所获得的pdcll2结晶结构还可以包含被限制在结构孔隙内的甲苯、乙苯和/或庚烷分子。
[0099]
在又另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的zn(ii)离子、以及作为抗衡阴离子的oac-,其中所述金属有机材料具有化学式(zn(oac)2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为znoacl2并且可以例如通过以下步骤而形成:在烘干的玻璃压力管中将l2的氯仿溶液添加到zn(oac)2·
2h2o的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热2天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡白色沉淀物形式的znoacl2,所述znoacl2具有球体的几何形状。或者,znoacl2可以通过以下步骤而形成:在烘干的玻璃压力管中将l2的氯仿溶液添加到zn(oac)2·
2h2o的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡白色沉淀物形式的znoacl2,所述znoacl2具有球体的几何形状。所获得的znoacl2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0100]
在另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的zn(ii)离子、以及作为抗衡阴离子的cl-,其中所述金属有机材料具有化学式(zncl2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为zncll2并且可以例如通过以下步骤而形成:在烘干的玻璃压力管中将l2的氯仿溶液添加到zncl2·
2h2o的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡白色沉淀物形式的zncll2,所述zncll2具有球体的几何形状。在一种替代性方法中,zncll2可以通过以下步骤而形成:在烘干的玻璃压力管中将l2的氯仿溶液添加到zncl2·
2h2o的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热2天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡白色沉淀物形式的zncll2,所述zncll2具有球体的几何形状。在另一种替代性方法中,zncll2可以通过以下步骤而形成:在烘干的玻璃压力管中将l2的氯仿溶液添加到zncl2·
2h2o的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热3天;然后在9小时至10小时内逐步冷却到室温,从而获得呈淡白色沉淀物形式的zncll2,所述zncll2具有球体的几何形状。所获得的zncll2结晶
结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0101]
在再另一个具体的实施方案中,本发明的金属有机材料包含各自是配体l2的配体、在结构上与所述配体的氮原子配位的zn(ii)离子、以及作为抗衡阴离子的br-,其中所述金属有机材料具有化学式(znbr2n2c
26.5h20
)n,其中n是至少4的整数,并且三维结晶微观结构或亚微结构任选地还包含溶剂分子。这种材料在本文被标示为znbrl2并且可以例如通过以下步骤而形成:在烘干的玻璃压力管中将l2的氯仿溶液添加到znbr2的dmf溶液中,然后将所述玻璃压力管密封;在105℃在没有搅拌的情况下并且在避光的情况下加热2天;然后在9小时至10小时内逐步冷却到室温,从而获得呈白色沉淀物形式的znbrl2,所述znbrl2具有球体的几何形状。所获得的znbrl2结晶结构还可以包含被限制在结构孔隙内的氯仿和/或dmf分子。
[0102]
在另一个方面,本发明涉及一种用于制备如上文所限定的金属有机材料的方法,所述方法包括以下步骤:(i)在压力容器中提供(a)金属盐的有机溶液或悬浮液,所述金属盐在本文被称为“金属组分”,由阴离子和所述金属离子组成;以及(b)所述配体的有机溶液或悬浮液,所述配体在本文被称为“有机组分”;(ii)将所述压力容器密封并且将所述压力容器在避光的情况下并且在没有搅拌的情况下保持一段时间,例如约1天、2天、3天、4天、5天、6天、7天、8天、9天或10天或更长时间,从而使所述金属离子与所述配体反应以获得呈沉淀物形式的所述金属有机材料;以及(iii)收集所述沉淀物。应当指出的是,除非另有说明,否则在说明和权利要求书中描述具体值的情况下,术语“约”意指应当假定所述具体值有可接受的误差范围,例如最多5%或10%。
[0103]
如本文所用的术语“压力容器”指的是被设计成在显著不同于环境压力的压力下容纳液体或气体的封闭容器。压力容器在理论上可以是几乎任何形状,例如具有半球形或碟形(准球形)的端盖,即头部的圆柱体,并且可以由任何合适的复合材料制成。在一个实施方案中,本发明的方法中所利用的压力容器是玻璃压力容器,更特别是本文所述的研究中所用的玻璃压力管。
[0104]
在某些实施方案中,在将容纳金属盐溶液/悬浮液和配体溶液/悬浮液的压力容器加热到60℃至120℃范围内的温度,例如60℃至70℃、70℃至80℃、80℃至90℃、90℃至100℃、100℃至105℃、105℃至110℃、110℃至115℃、或115℃至120℃,持续整个所述时间段或其一部分,然后逐步冷却所述压力容器的同时进行本发明的方法的步骤(ii)。如本文所示,在一些情况下,步骤(ii)包括将所述压力容器密封并且将所述压力容器在避光的情况下并且在没有搅拌的情况下保持一段时间,同时加热到如上文所限定的温度,持续整个时间段,然后在步骤(iii)之前逐步冷却例如到室温。或者,步骤(ii)可以包括将所述压力容器密封并且将所述压力容器在避光的情况下并且在没有搅拌的情况下保持一段时间,同时如上文所限定进行加热,持续所述时间段的一部分,即在所述时间段开始时、在所述时间段期间、或在所述时间段结束时,然后逐步冷却例如到室温。
[0105]
在某些实施方案中,在惰性条件下,例如在氩气或n2下进行本发明的方法的步骤(i)和(ii)。
[0106]
其中溶解所述配体和金属盐的有机溶剂可以独立地是极性的或非极性的,其中所述配体在其中溶解它们的有机溶剂中的溶解度决定了所述有机组分将呈溶液还是悬浮液的形式,并且所述金属盐在其中溶解它的有机溶剂中的溶解度决定了所述金属组分将呈溶
液还是悬浮液的形式。有机溶剂的非限制性实例包括氯仿、二甲基甲酰胺(dmf)、烷醇,如甲醇、乙醇、异丙醇、正丁醇、仲丁醇、异丁醇、戊醇和己醇;dmso、乙腈、乙二醇、甲苯、苯、乙苯、乙醚(ether/diethyl ether)、以及烷烃,如戊烷、己烷、环己烷、庚烷、环庚烷、辛烷、环辛烷、壬烷、癸烷以及十氢化萘。
[0107]
如本文所示,在金属盐在所用的有机溶剂中的溶解度不佳,然而需要金属盐的有机溶液,而不是悬浮液的情况下,可以添加能够与金属原子形成配位络合物,即金属络合物,从而提高所述金属盐在所述有机溶剂中的溶解度的化合物。在本文所举例说明的具体的这样的情况下,将其中pd原子与1,5-环辛二烯(cod)或苯甲腈(phcn)配位的pdcl2(即分别是pd(cod)cl2或pd(phcn)2cl2)溶解在甲苯或乙苯中以获得pdcl2的甲苯溶液或乙苯溶液而不是悬浮液,。
[0108]
根据本发明的方法,由于在步骤(ii)期间在压力容器中进行的反应而获得金属有机材料。如本文明确所示,包含相同的配体和金属离子,但是具有不同的三维结晶结构,从而具有潜在不同的物理特性和化学特性的金属有机材料是根据反应组分以及反应条件而获得的,所述反应组分例如阴离子和其中溶解所述配体和金属盐的有机溶剂,所述反应条件例如在步骤(ii)中使所述有机组分与金属组分反应的时间段、进行反应的温度或反应的热曲线(在所述反应包括在所述时间段的一部分内进行加热的情况下)、以及冷却速率(在所述反应包括在整个所述时间段或其一部分内进行加热的情况下)。
[0109]
通过本发明的方法获得的金属有机材料包含金属离子,优选地过渡金属离子,所述金属离子与各自具有通式i的有机配体配位以形成一维结构、二维结构或三维结构,其中所选择的具体的金属离子和有机配体决定了所述材料的结构并且因此决定了物理特性和化学特性。在某些实施方案中,所述金属有机材料具有三维结晶微观结构或亚微结构,所述结构可以具有如上文所限定的特定的几何形状。应当了解的是,虽然使用不同的反应组分和/或在不同的反应条件下所获得的结晶微观结构和亚微结构可以具有不同的几何形状,但是如上文所限定的利用特定的反应组分并且在特定的反应条件下进行的制备方法产生了一群具有均一的几何形状的三维晶体结构。
[0110]
在某些实施方案中,通过本发明的方法所获得的三维微观结构和亚微结构的几何形状因此受步骤(ii)中的反应组分和/或反应条件或参数的影响,其中所述反应组分是例如所述金属离子、阴离子、以及有机溶剂中的一种或多种,并且所述反应条件或参数是例如所述温度、时间段以及冷却速率中的一种或多种。
[0111]
本技术的mof可用作用于气体吸附的工艺中的吸附剂,例如h2、co、co2或甲烷吸附、或气体分离和/或纯化,例如将co2和甲烷分离。
[0112]
在又另一个方面,本发明因此涉及如上文所限定的金属有机材料用作用于气体吸附或气体分离的工艺中的吸附剂的用途。
[0113]
在再另一个方面,本发明涉及一种用于气体吸附或气体分离的方法,所述方法是通过使所述气体吸附到吸附剂上来实现的,其中改进之处在于所述吸附剂是如上文所限定的金属有机材料。
[0114]
现在将通过以下非限制性实施例来说明本发明。实施例材料和方法
[0115]
一般方法:将玻璃压力管通过在食人鱼溶液(piranha solution)(7:3v/v的h2so4/30%h2o2)中浸泡10分钟并且在去离子(di)水中浸泡来清洁,然后在130℃干燥12小时。警告:食人鱼是一种非常危险的氧化剂并且应当使用适当的个人防护加以小心处理。
[0116]
透射式电子显微镜法(tem):使用装备有电荷耦合器件照相机(2k
×
2k gatan ultrascan 1000)的在120kv下操作的philips cm-120仪器进行tem成像。通过将一滴5μl的反应混合物放置在聚乙烯醇缩甲醛树脂(formvar)/碳、400筛目cu网上并且在10秒之后吸去来制备tem样品。由于样品的光束敏感性,因此在低剂量条件下进行tem成像和saed测量。使用edax eds系统进行元素分析。
[0117]
扫描电子显微镜法(sem):使用hrsem ultra-55 zeiss和hrsem supra-55vp zeiss仪器在3kv的eht电压下进行sem测量。通过将一滴反应混合物或分离的mof的dmf悬浮液放置在硅衬底上,将所述硅衬底在空气下干燥来制备sem样品。
[0118]
ftir和nmr光谱法:使用nicolet 460单光束ft-ir获得红外光谱。在300mhz bruker nmr光谱仪上进行1h和
13
c{1h}nmr测量。
[0119]
原子力显微镜(afm):在p47 solver afm(俄国泽廖诺格勒的nt-mdt公司(nt-mdt,zelenograd,russia))上使用ac240探针(奥林帕斯公司(olympus))以断续接触模式,以及用multimode 8afm(加利福尼亚州圣巴巴拉的布鲁克公司(bruker,santa barbara,ca))进行afm表面特征成像。以“峰力定量机械映射(pf-qnm)”模式操作后一种系统,所述模式使得能够与表面特征图像同时采集弹性模量。对于这种测量,使用bruker rtespa探针。由sader法(sader等,1999)测定的弹簧常数是80n/m。弹性模量源自于在每一个像素所采集的力曲线,并且依赖于一些校准(悬臂偏转灵敏度、弹簧常数、尖端半径),将这些校准输入到dmt分析中(tabor,1977)。变形是约5nm-10nm,因此对尖端和样品表面条件是非常敏感的,所述表面条件在扫描过程期间可以改变有效尖端半径。在模量测量中的估计不确定性是30%。如同在sem的情况下那样,在硅衬底上制备样品。
[0120]
快速热处理(rtp)和热重分析(tga):在快速热退火炉(rapid thermal annealer)上,在10%h2/n2流下或在真空下,在不同的温度(200℃-600℃)将rtp进行5分钟。如同在sem的情况下那样,制备样品。在sdt q600 v8.3 build 101仪器上使用氧化铝样品盘在n2流下进行tga。
[0121]
磁性测量:使用squid磁强计mpms xl测量分离的样品的磁特性。将样品称重并且放置在明胶胶囊中并且从室温冷却到2k而没有施加任何外磁场(zfc)和强度1000oe的内磁场(fc)。在施加的外磁场(h=1000oe)下从2k加热到300k期间测量磁矩的温度依赖性。使用估计化学式nicll1(nicl2n2c
26.5h16
)n和nibrl2(nibr2n2c
26.5h20
)n将磁化率对温度的依赖性归一化。在包括dmf(0.5-4个分子/ni)的情况下,这种依赖性是相似的。发现zfc依赖性和fc依赖性是重叠的(图20a、21)。韦斯方程(weiss equation)定义了磁化率的温度依赖性并且可以表示为χ=χ
0 c/(t-θ)
,其中c是居里常数(curie constant),χ0是非温度依赖性参数并且θ对应于韦斯参数。
[0122]
气体吸收研究:在压力组成等温线(pci)仪器(美国的创新材料公司(advanced materials corporation,usa))上进行气体吸附研究。在氩气下将约50mg的化合物,例如nicll1加入手套箱内的样品室中。然后将样品室与真空管线连接。通过缓慢加热到120℃(对于ch4吸附)以及70℃、100℃和120℃(对于h2吸附)并且在这些温度下保持4小时,继而冷
却到室温来活化样品。在测量期间保持样品室的温度恒定。使用希沃特原理(sieverts principle)测定气体吸收。通过使用氦气(10巴)进行气体测比重术来测量样品的密度。
[0123]
用于合成mof的一般程序:在玻璃压力管中,将多吡啶配体l1(schilling等,2011)或l2(thompson等,1997)(6.8μmol)的chcl3溶液(1.0ml)添加到以下相应金属盐(对于1当量来说,是6.8μmol;并且对于2当量来说,是13.6μmol)的dmf溶液(3.0ml)中:nicl2、nibr2、cucl2、cubr2以及cu(no3)2。然后,将所述管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热5天,继而在9小时至10小时内以10℃/小时逐步受控冷却到室温。这产生沉淀物(对于基于ni的mof来说,是淡绿色的,并且对于基于cu的mof来说,是深绿色的)。通过将反应混合物以5000rpm离心约10分钟并且滗析母液而以接近定量产率(》95%)收集mof。研究1:均匀微观结构化的结晶镍有机配位聚合物
[0124]
通过如所述的差示溶剂热合成获得配位聚合物的微小尺寸的砖状结构。在密封的压力管中在暗处将1当量的nicl2·
6h2o的dmf溶液与0.5当量的配体l1的氯仿溶液一起在105℃加热5天,然后在9小时至10小时内进行缓慢冷却。在混合这两种溶液时立即形成白色沉淀物,并且使所述白色沉淀物在不受干扰的情况下在所提到的条件下静置和沉降。在冷却到室温之后,通过离心收集淡绿色沉淀物。
[0125]
nicll1的sem图像揭示了砖状结构,并且对来自nicll1的代表性样品的三百个随机选择的砖状结构进行的统计分析确定尺寸分布的合理均匀度(图1),大部分的结构拟合于2.5μm-2.7μm的长度、1.2μm-1.4μm的宽度以及200nm-300nm的厚度。tem图像显示所述材料的结晶性质;可以经由tem成像和saed识别晶格面的存在(图2);并且发现最长的对称轴是沿着晶体的长度。
[0126]
afm研究揭示了与在电子显微照片中所看到的相同的形状以及所述材料的表观表面粗糙度。还在tem显微照片中所观测到的碎片似乎是无序材料,高度在数十纳米至超过一百纳米的范围。雏晶的上表面总体上是平坦的,尽管可以观测到分散的凹陷以及出现在表面的部分上的新层(图3)。
[0127]
对这些纳米结构进行的弹性模量的测量给出了2-12吉帕斯卡(gpa)的值。这些值在对于有机晶体所观测到的那些值的范围内(roberts等,1991)。在表面特征图像同时测量弹性模量,因此值在像素水平上与表面特征相关。图2示出了模量和相应表面特征的图像。在硅衬底上的模量信号对于这一探针来说在高值处达到饱和。对于这些阶梯式特征,更高区域的模量是更低区域的约一半。这一观测结果与如下的前提是一致的,即雏晶在生长期间经受抛光过程,其中更高的不太有序的区域随时间被去除。这些不太有序的区域预期是可更大程度变形的并且因此显示出更低的模量值(图4)。
[0128]
nicll1的ft-ir谱(kbr压片)显示出与游离配体l1相比20cm-1
的偏移(图5)。不出所料的是,浓酸消化了整个系统,并且在用et3n中和之后用氯仿萃取的所得混合物的1h nmr与配体l1完全匹配(图6),并且对粗混合物进行的质谱分析揭示了配体l1和nicl2的存在。maldi-tof分析还确认在络合物nicll1中配体是完整的。通过纳米探针x射线eds确认微砖的元素组成,揭示了ni、cl以及n原子的存在,并且发现ni/n比率是0.44(图7)并且所述值与每个ni中心包含两个吡啶的完全形成的配位饱和网络的形成(kaminker等,2010;choudhury等,2010)是接近一致的,但是其它配位模式可能并不被完全排除在外。
[0129]
这些明确限定的矩形结构的形成可以被视为面或轴向限制生长机制的影响,但是
对这些均匀结构的形成的机制了解仍是初步的并且可以预期遵循了一种规则的配位过程,继而是热引发的通过使单个构建块熔合所引起的紧密堆积。在反应过程期间进行的与时间有关的sem分析揭示了最终所观测到的结构的形成的推测性的,但是合理的机制。如所预期,配体l1在与ni盐溶液混合时立即形成相应的配位聚合物,所述配位聚合物具有不均匀的棒状和非常小的块状结构(图8,上图),这进而在加热时在压力下熔合以形成砖状微观结构(图8,中图)。在任何时间点时溶剂混合物的影响均可能不能被完全排除在外,这是因为配位聚合物即刻沉淀并且因此可以解释经由附聚所引起的表面张力减小。随时间推移并且在高温的影响下,这些结构的表面在很大程度上变得平滑,从而产生具有一定的表观表面粗糙度的平坦面(图8,下图)。因此,所提出的工作机制涉及配位/成核作用,继而是聚集/低聚、熔合/生长以及退火。mirkin和同事已经提出了在朝格尔碱(troger's base)前体和zn(ii)聚合期间形成完美的微球体的类似机制(spokoyn等,2009;oh和mirkin,2005;jeon等,2009)。
[0130]
发现由此所获得的微观部分在包括水在内的所有常用溶剂中是不可溶的并且发现其在空气中在室温下在大部分的常用溶剂中是稳定的。所述结构的这种稳定性可以通过在暗处悬浮在水、dmf以及这两者的混合物中的固体材料在2个月后所拍摄的sem图像来确定(图11)。
[0131]
发现配位聚合物的最终结构取决于一些参数,包括所使用的技术、试剂的摩尔比、溶剂系统、反应时间以及温度。已经提到的是,将配体l1的chcl3溶液与nicl2·
6h2o的dmf溶液在室温混合得到了不均匀的棒状和非常小块状结构。以1:1或1:2的摩尔比小心地使配体l1的chcl3溶液在ni盐的dmf溶液下分层在允许在暗处静置过夜时得到了淡绿色沉淀物,并且代表性样品的sem图像显示更长并且更均匀的棒状物和一定量的没有明确限定的结构的大块材料的混合物。在压力管中将这种混合物(在约4天内完全扩散之后)加热到80℃-105℃,持续3天至5天使在sem下所观测到的结构发生剧烈的变化,这表明由此所获得的更长的棒状物是不稳定的并且在热处理下进行随机的熔融和熔合,这是因为大块材料连同棒状物一起似乎已经聚集以形成某种形式的团块。
[0132]
尽管改变配体和金属的摩尔比没有太大地影响分层技术的结果,但是发现在热方案下对结构特征有实质影响。在相同的溶剂系统中在相同的条件下,但是以1:1的比率将配体和金属加热会产生具有更宽的尺寸分布的更小的并且不同的砖状结构(图12)。
[0133]
还发现用于反应的溶剂对在电子显微镜下所观测到的最终结构有显著的影响。单独使用dmf作为溶剂,即使配体l1是不可溶的(图13,图b)也会使得砖状结构完全丧失,其中使用dmf/chcl3的1:2混合物代替1:3混合物会引起相对更明确的结构形成,但是没有观测到真正的砖状部分(图13,图c)。这可以被解释为质子在chcl3中的酸性性质,所述chcl3能够施加与吡啶的氢键键合相互作用,从而干扰孤对的配位能力。将0.25ml至0.5ml的水添加到1:3的dmf/chcl3混合物中完全破坏了所述系统(图13,图d)并且这些结果也可能强调了溶剂极性对这些明确限定的矩形结构的形成的影响。可以指出的是,在除了添加少量水之外所有的情况下,配位聚合物的沉淀均是瞬时的并且具有在标准条件下所获得的白色不溶性材料的物理外观。
[0134]
材料的热行为是配位聚合物的又另一个受关注的方面。在10%h2/n2流下对砖状ni(ii)配位聚合物进行rtp确定了所述结构对于约300℃的温度是稳定的(图14、15)。此外,发
现掺杂硼的硅表面会对这些结构诱导更好的稳定性并且可以指出的是,平躺在表面上的微砖在甚至更高的温度下仍保持它们的形状。当在高于400℃下接受处理时,发现熔融的结构点缀有金属镍的纳米粒子(约20nm-25nm)。
[0135]
另一个重要的观测结果是如果在真空下接受热处理的话,所述结构似乎稳定得多(图16),即使是在所述事件期间仍发现发生金属纳米粒子点缀。
[0136]
对砖状结构进行的tga允许我们确认在晶格内部作为配位溶剂的dmf的存在,这是因为在约150℃观测到重量减轻(图17)。如在快速热退火之后所发现,所述结构在这一温度下是相当稳定的并且它仅可能是在dmf的这一特征性沸点下气化的溶剂并且因此证实了所观测到的重量减轻是合理的。
[0137]
金属有机框架和配位聚合物有作为储气材料的受关注的应用(manson等,2014;adisa等,2012)。随压力的变化对nicll1的气体吸附特性进行研究并且对预活化的样品进行的实验证实合理量的甲烷(在环境温度或0℃下约7.5重量%以及在-78.5℃和约35atm压力下11.7重量%)和氢气(在约35atm压力下约1.75重量%)吸附(图18、19(h2吸附-解吸等温线))。此外,在测量期间,样品在吸附操作与解吸操作之间表现出非常少的滞后。还使用同一技术来测定物质的密度并且发现是约0.687g/cc。
[0138]
squid测量揭示了nicll1和nibrl2这两者的顺磁行为。发现zfc依赖性和fc依赖性是重叠的(图20a、21)。这些磁特性与金属中心的接近四面体或八面体配位几何形状是一致的(bridgeman,2008)。
[0139]
已经经由包括电子衍射、粉末xrd以及同步辐射衍射在内的各种技术进行尝试来解答nicll1的晶体结构,但是迄今为止仍是不成功的,这部分地是因为雏晶的尺寸(这使得难以分离单晶以及具有雏晶尺寸范围内的相容的束线)并且部分地是因为所观测到的衍射图的低强度(特别是在电子衍射中)。尽管如此,磁特性使可能的几何形状变得明朗,顺磁行为在很大程度上忽略了正方形平面ni(ii)络合物的可能性,从而留下了四面体或八面体几何形状的选项。已知的是,在正方形平面络合物中,配体以完全电子配对作为代价施加与金属的非常强的σ相互作用,从而使得σ*轨道未占用,产生抗磁行为(bridgeman,2008)。研究2:金属有机框架的拓扑控制——从矩形砖状物到星形和互穿多面体
[0140]
在这一研究中,我们证实对mof的结构均匀度和多样性的控制。更具体地说,我们引入了具有窄的尺寸分布的一系列三维(亚微结构化)微观结构化mof的组装体以及对它们的拓扑结构加以优良的控制。通过系统地改变以下各项展现出不同的结构,从细长的六边形和矩形棱柱到星形和互穿多面体:(i)金属中心;(ii)阴离子;(iii)有机配体;以及(iv)反应条件,即溶剂、温度、以及有氧或无氧。举例来说,使用ni(ii)盐产生了不同的多面体拓扑结构,相反,cu(ii)前体形成了互穿和/或星形多面体。这些金属有机结构是非常少见的(masoomi和morsali,2013)。经由在没有使用表面活性剂或外部调节剂(例如吡啶、十六烷基三甲基溴化铵)的情况下进行溶剂热合成实现了我们的材料在形状和尺寸方面的均匀度(sindoro等,2014;gao等,2014;ranft等,2013;guo等,2012;cho等,2008)。对mof的形成进行的后续的电子显微镜法研究揭示了一系列复杂的反应。对于基于ni的mof,观测到两种类型的生长过程,所述生长过程涉及成核作用和抛光,而熔合过程在基于cu的mof的形成中发挥了主要的作用。
[0141]
为了实现和合理化对金属有机微观结构的拓扑控制,需要如下的配体-金属-阴离
子组合:(i)通过互连的四面体节点形成稳健的和延伸的三维网络,从而允许产生钻石形网络(batten,2001);以及(ii)产生并有溶剂分子的极高的永久性微孔隙和/或通道以使微观结构稳定。因此,我们使用了两种有机配体l1和l2以及可商购获得的ni(ii)盐和cu(ii)盐。这些四面体配体具有刚性,具有完全的td对称性和四个金属离子结合位点。这一组合确保了稳健的、多孔的并且延伸的三维网络的形成(lu等,chem.soc.rev.,doi:10.1039/c4cs00003j)。ni(ii)盐和cu(ii)盐对吡啶基配体具有高亲和力(tomasik等,2008;hasenknopf等,1996),尽管如此,金属-n键强度允许动态结构在高温下进行重排而形成热力学产物(kaminker等,2011)。通过使用具有不同的配位要求的金属来证实这一研究的范围。此外,已经证实了阴离子在我们的微观结构的形成中的主要作用。
[0142]
在典型的实验中,将金属盐的dmf溶液与0.5当量或1当量的l1或l2的氯仿溶液混合并且在玻璃压力管中在105℃在避光的情况下加热。在4天至5天之后,在9小时至10小时内将反应混合物逐步冷却并且通过离心定量地收集微观结构。已经通过电子显微镜分析、xrd以及afm对结晶微观结构进行表征。已经通过红外(ir)光谱法、磁性测量、以及气体吸附获得了在分子水平上的信息。还已经测试了镍结构的热稳定性。
[0143]
sem和tem成像揭示了nicl2和l1或nibr2和l2的2:1比率的组合分别产生了单分散结构(nicll1和nibrl2;图22和23)。尽管这两种mof均具有规则的六边形拓扑结构,但是nicll1形成了明显细长的六边形,这还可以通过光学显微镜法来观测(图10)。这些观测结果证实了有机配体(即l1:c≡c对比l2:c=c)和阴离子(cl、br)中的微小的结构差异是关键因素,所述关键因素可以用于在微观水平上调整这些mof的拓扑结构,而保持高水平的均匀度。此外,可以使用金属与配体和溶剂的比率来控制mof拓扑结构。举例来说,以1:1的比率使用nicl2和l1会产生更小的六边形拓扑结构,而改变氯仿的含量会产生细长的结构(图24)。向反应物中添加水会引起结构变形(图24,图f)。
[0144]
对nicll1和nibrl2进行的afm测量确认了拓扑结构并且允许精确测量结构高度(图3)以及确定机械特性(材料和方法可以补充材料的形式在science online上获得)。通过afm纳米压痕所测量的nicll1的弹性模量是5gpa-6 gpa,这类似于对于有机晶体所报道的值(roberts等,1991)。这两种mof的结晶性质明确地通过saed来证实(图25)。对nicll1进行的xrd测量还表明了有序结构的形成(图9)。定性地通过x射线eds确认了mof的元素组成,显示出对应于所有特征原子(氮、金属以及阴离子)的峰(图7和26)。通过ft-ir光谱法确认了配体的存在,显示出对应于nicll1和nibrl2的配体框架的峰,所述峰与游离配体相比有所偏移(图5)。l1和l2的分子结构不可能在这些镍盐的存在下受溶剂热条件的影响。这种假设是通过在强酸性条件(ph值《1)下溶解mof,随后对有机组分进行分离和表征来验证的。1h和
13
c{1h}nmr光谱法(图6和27)和质谱分析(esi-ms和maldi-tof)确认了配体稳定性。squid测量揭示了nicll1和nibrl2这两者的顺磁行为。发现zfc依赖性和fc依赖性是重叠的(图20a、21)。这些磁特性与金属中心的接近四面体或八面体配位几何形状是一致的(bridgeman,2008)。
[0145]
分离的nicll1和nibrl2在室温下在暗处在至少一年内保持空气中的稳定性。将这些mof在dmf或水中浸泡数月没有引起它们的微观结构方面的任何可观测到的变化。nicll1的tga显示在约86℃有3.4%的相对小的重量减轻,这对应于chcl3的丧失(图17)。在1000℃有30%的重量减轻。在10%h2/n2流下对nicll1和nibrl2进行的rtp和后续的sem分析表明在
200℃下保持所述结构。在更高的温度下观测到nicll1和nibrl2这两者明显的变形(图28和29)。发现所述结构在≥400℃下点缀有金属纳米粒子nicll1在真空下的拓扑稳定性甚至更高,这表明热稳定性受h2影响。
[0146]
通过气体吸附分析来证实nicll1的孔隙度。将nicll1在120℃在高真空下活化数小时以评价它对天然气(ch4)的吸附/释放效率。在35atm的压力下,ch4吸附在0℃-20℃是7.5重量%,并且在-78.5℃是11.7重量%。吸附操作与解吸操作之间的滞后是可忽略的,这确认了微孔性和ch4吸收的可逆性(图18)。气体测比重术表明了0.687g/cc的密度。nicll1的ch4吸附能力(75cm
3 stp/cm3)在cof-10、cd2(azpy)3no3、co2(4,4'-bpy)2(no3)4、cu2(pia)2(no3)4以及可商购获得的basolite a520的吸附能力的范围内(manson等,2014;adisa等,2012)。
[0147]
使用cu盐产生了具有显著不同的结构的mof。使用cucl2和l2获得了不均匀结构化的mof(图30)。然而,使cubr2与l2反应引起了两个互穿四面体(星型八面体)的形成,这可以被描述为大卫之星(star of david)的三维延伸(cubrl2;图31)。反应条件与用于形成nicll1和nibrl2的反应条件是相同的(图22)。通过xrd明确地确认基于cu的mof的结晶性质;获得与nicll1相似的粉末xrd图。
[0148]
阴离子的性质和金属与配体的比率对于明确限定的基于cu的mof的形成来说也是关键参数。使用cu(no3)2和1:2(金属:l2)比率产生了不明确的结构,与对于1:1比率所获得的更高的均匀度形成对比。后者产生了花状拓扑结构(cu(no3)l2;图32和33)。有趣的是,在严格排除空气和使用无水溶剂的情况下进行这一反应引起了矩形棱柱的形成,所述矩形棱柱具有3.65
±
0.95μm的平均长度和0.675
±
0.09μm的宽度(cu(no3)l2;图34)。与其它基于cu的mof对比,cu(no3)l2没有显示出任何互穿的证据。通过ft-ir光谱法确认l2的存在和配位,显示出对应于cubrl2和cu(no3)l2的配体框架的峰(图35)。在酸性条件下溶解基于cu的mof,以及后续通过nmr光谱法和质谱分析对l2进行的分离和表征确认了它的稳定性。发现这三种基于cu的mof与nicll1和nibrl2相比不太均匀;然而,它们仍具有共同的结构基序。基于cu的mof的均匀度较小可能是与它们更高的结构复杂性和可能结构的更大多样性相关的。
[0149]
mof的形成可能是组装过程的复杂级联的结果(spokoyny等,2009;oh和mirkin,2005)。对于基于ni的mof和基于cu的mof这两者,溶剂组成对于均匀结构的产生来说同样起关键性的作用(图24、36以及37)。改变dmf/chcl3比率和/或添加其它溶剂(phcn、dmso、水)会产生不同的组装体。
[0150]
对基于ni的mof和基于cu的mof的形成的电子显微镜研究进行扩展揭示了有趣的机制信息。与时间有关的分析证实了形成所获得的均匀结构的明显不同的途径。将nicl2盐和nibr2盐的溶液与相应配体(l1或l2)混合引起立即沉淀。显然,所述过程开始于配体与金属中心的配位作为第一成核步骤,如结晶和胶体合成中所常见的那样。对在混合后立即获取的nicll1等分试样进行的sem分析证实了细长的(针状)结构和立方体结构(《1μm;图38,图a)的混合物的形成。这种混合物的热分解引起了早期六边形结构的形成,其总体形状和尺寸类似于最终产物,但是具有粗糙的纹理和边缘(图38,图b)。连续加热5天得到抛光的nicll1(图23和38,图c)。由mirkin所报道的无定形的无限配位聚合物(icp)经受了类似于在此所观测到的结构抛光的退火(spokoyny等,2009;jeon等,2009)。粗糙的表面对于添加
更多的材料来说可能是理想的成核点。对于nibrl2的形成来说,不同的生长过程起作用。在混合的初始阶段,形成小并且均匀的雏晶(约55nm
×
27nm),所述雏晶与最终产物具有相同的拓扑结构(nibrl2;图23和39,图d)。在反应期间,它们的尺寸增大到差不多五倍(图38,图d-e)。对于这两种基于ni的mof,更高的温度和压力提高了纳米结构的平均尺寸并且减少了更小的纳米结构的数目。更小的结构的更高的表面能可能会促进它们溶解,从而产生新核(murray等,2001)。不同于在nicll1的情况下所观测到的抛光过程,对于nibrl2来说,不同的机制起作用,所述机制涉及通过向核添加材料所引起的正方晶生长而在形成过程中保持相同的基本形状(类似于奥氏熟化(ostwald ripening))。
[0151]
对基于cu的mof的生长进行的与时间有关的sem分析揭示了涉及多种中间结构的相当复杂的序列。将cubr2溶液与l2混合在室温下产生了不均匀的片状结构(图39,图a),在加热时在1.5天之后,所述结构转变成侧向熔合的球体(直径=650
±
50nm,图39,图b)。在连续加热后,观测到大得多的钻石状结构(图39,图c)以及其熔合结构。一些球形结构保留,然而更小(图39,图c,插图)。有趣的是,在3.5天之后,主要呈现出金字塔结构,这最有可能是由熔合和成核作用的组合而形成的(图39,图d、d')。图39图d的插图明确地示出了穿透双型结构。最初的金字塔形状是由钻石状结构的熔合而形成的(图39,图c、d),并且它们的小平面随后充当成核点以得到在图39图d'中所看到的在动力学上复杂的产物。进一步加热产生了在热力学上稳健的cubrl2(图32和39,图e),其具有双晶的外观。
[0152]
cu(no3)l2的形成开始于不均匀的片状结构的形成,所述结构类似于对于cubrl2所观测到的结构(图40,图a,插图)。在加热一天之后,形成不规则的矩形棱柱(图40,图a),在2.5天之后其转变成互穿结构(图40,图b)。在继续加热后,这些明显的带螺纹的系统进行另一熔合过程以提供花状拓扑结构(图40,图c、d)。
[0153]
我们的观测结果证实了具有均匀的尺寸分布的金属有机微晶的形成可以容易地通过溶剂热合成来实现。他人已经使用溶剂热方法主要在分子水平上实现结构的修饰(stock和biswas,2011)。此外,经由系统化学修饰所引起的晶体堆积变化对于许多有机材料和其它材料来说是已知的(stock和biswas,2011;zhao等,2011;smulders等,2013;wang等,2013;shirman等,2008)。然而,这样的获得均匀微晶的方法是罕见的(masoomi和morsali,2013;ban等,2013)。值得注意的是,改变分子内结构(即c≡c对比c=c;cl对比br;ni对比cu)对在此所报道的mof的形成、均匀度以及拓扑结构有这样显著的影响。我们在(亚微米)微米水平上获得均匀度的方法对金属在元素周期表中的位置以及反应时间是敏感的。举例来说,我们先前已经证实了pd(ii)盐与l2反应会引起配位聚合物纳米管的形成(kaminker等,2011)。这些纳米管的结构特征和尺寸具有组装依赖性,如由aida等(zhang等,2009)所示。在本研究中,最初观测到(亚微)微观结构的混合物,所述混合物逐步转变成均匀结构化的晶体。尽管这一研究并没有试图对预先决定这些微观结构的拓扑结构的因素进行表征,但是引出了定制设计的拓扑结构的可能性。考虑到在这一研究中所考虑的参数的范围,可以预期的是,mof的形状特异性和尺寸均匀性的可能性是一个广泛和普遍的现象。研究3:各种金属有机框架
[0154]
在本文以下所述的一系列实验中,已经使用上文所述的一般程序制备了各种mof,其中利用了不同的金属盐和反应条件。
(2.8mg,7.8μmol)的dmf溶液(3.0ml)下方分层,将所述玻璃管密封并且在暗处保持10天,然后在60℃在没有搅拌的情况下并且在避光的情况下再加热2天,继而后续冷却到室温,从而产生淡蓝色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图49)。
[0164]
在n2气氛下在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的无水chcl3溶液(1.0ml)添加到cu(no3)2·
3h2o(1.65mg,6.8μmol)的无水dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热6天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生淡绿色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图50)。
[0165]
在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的chcl3溶液(1.0ml)添加到zn(oac)2·
2h2o(3mg,13.6μmol)的dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热2天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生淡白色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图51)。
[0166]
在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的chcl3溶液(1.0ml)添加到zn(oac)2·
2h2o(3mg,13.6μmol)的dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热3天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生淡白色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图52)。
[0167]
在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的chcl3溶液(1.0ml)添加到zncl2·
2h2o(0.92mg,6.8μmol)的dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热3天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生淡白色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图53)。
[0168]
在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的chcl3溶液(1.0ml)添加到zncl2·
2h2o(1.86mg,13.6μmol)的dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热2天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生淡白色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图54)。
[0169]
在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的chcl3溶液(1.0ml)添加到zncl2·
2h2o(1.86mg,13.6μmol)的dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热3天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生淡白色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图55)。
[0170]
在烘干的玻璃压力管中,将l2(5mg,6.8μmol)的chcl3溶液(1.0ml)添加到znbr2(3.1mg,13.6μmol)的dmf溶液(3.0ml)中,将所述玻璃压力管密封并且在105℃在没有搅拌的情况下并且在避光的情况下加热2天,继而后续在9小时至10小时内以10℃/小时的速率受控冷却到室温,从而产生白色沉淀物并且通过将反应混合物以5000rpm离心约10分钟并且滗析母液来收集所述沉淀物(图56)。
附录方案1:多吡啶配体l1-l4的化学结构l4的化学结构参考文献adisa,o.o.,cox,b.j.,hill,j.m.,nanoscale,2012,4,3295-3307。bailar,j.c.,jr prep.inorg.react.,1964,1,1-57。ban,y.,li,y.,liu,x.,peng,y.,yang,w.,microporous and mesoporous materials,2013,173,29-36。batten,s.r.,crystenggcomm,2001,18,1-7。biradha,k.,ramanan,a.,vittal,j.j.,crystal growth and design,2009,9,2969-2970。bridgeman,a.j.,dalton trans.2008,1989-1992。carn
é‑sá
nchez,a.,imaz,i.,stylianou,k.c.,maspoch,d.,chem.eur.j.,2014,20,5192-5201。chen,b.,xiang,s.,qian,g.,acc.chem.res.,2010,43,1115-1124。
cho,w.,lee,h.j,;oh,m.,j.am.chem.soc.,2008,130,16943-16946。choudhury,l.,kaminker,r.,motiei,l.,de ruiter,g.,morozov,m.,lupo,f.,gulino,a.,van der boom,m.e.,j.am.chem.soc.,2010,132,9295。cook,t.r.,yang,r.y.,stang,p.j.,chem.rev.,2013,113,734-777。evans,o.r.,lin,w.,acc.chem.res.,2002,35,511。furukawa,h.,cordova,k.e.,o'keeffe,m.,yaghi,o.m.,science,2013,341,1230444。gao,j.,ye,k.,yang,l.,xiong,w.w.,ye,l.,wang,y.,zhang,q.,inorg.chem.,2014,53,691-693。gu,x.w.,loynachan,c.n.,wu,z.,zhang,y.w.,srolovitz,d.j.,greer,j.r.,nano lett.,2012,12,6385-6392。guo,y.n.,li,y.,zhi,b.,zhang,d.,liua,y.,huo,q.,rsc adv.,2012,2,5424-5429。hasenknopf,b.,lehn,j.m.,baum,g.,fenske,d.,proc.natl.acad.sci.usa,1996,93,1397-1400。jeon,y.m.,armatas,g.s.,kim,d.,kanatzidis,m.g.,mirkin,c.a.,small,2009,5,46-50。jung,s.,oh,m.,angew.chem.,int.ed.2008,47,2049-2051。kaminker,r.,motiei,l.,gulino,a.,fragal,i.,shimon,l.j.w.,evmenenko,g.,dutta,p.,iron,m.a.,van der boom,m.e.,j.am.chem.soc.,2010,132,14554。kaminker,r.,popovitz-biro,r.,van der boom,m.e.,angew.chem,int.ed.,2011,50,3224-3226。kitagawa,s.,kitaura,r.,noro,s.,angew.chem.,int.ed.2004,43,2334。kittel,c.,《固态物理学的入门》(introduction to solid state physics).john wiley and sons.,1996。kondo,m.,okubo,t.,asami,a.,noro,s.-i.,yoshitomi,t.,kitagawa,s.,ishii,t.,matsuzaka,h.,seki,k.,angew.chem.int.ed.1999,38,140-143。lei,b.f.,li,b.,zhang,h.r.,zhang,l.m.,li,w.l.,j.phys.chem.c,2007,111,11291。li,w.,doblinger,m.,vaneski,a.,rogach,a.l.,jackel,f.,feldmann,j.,j.mat.chem.,2011,21,17946-17952。liu,k.,you,h.,jia,g.,zheng,y.,huang,y.,song,y.,yang,m.,zhang,l.,zhang,h.,crystal growth and design,2010,10,790-797。long,j.r.,yaghi,o.m.,chem.soc.rev.,2009,38,1213-1214。lu,y.,cao,h.,zhang,s.,zhang,x.,j.mater.chem.,2011,21,8633-8639。lu,w.,wei,z.,gu,z.y.,liu,t.f.,park,j.,park,j.,tian,j.,zhang,m.,zhang,q.,gentle iii,t.,bosch,m.,zhou,h.c.,chem.soc.rev.,doi:10.1039/c4cs00003j(advance article)。manson,j.a.,veenstra,m.,long,j.r.,chem.sci.,2014,5,32-51。
masoomi,m.y.,morsali,a.,rsc adv.,2013,3,19191-19218。murray,c.b.,sun,s.,daschler,w.,doyle,h.,betley,t.a.,kagen,c.r.,ibm.j.res.&dev.,2001,45,47-56。ni,z.,masel,r.i.,j.am.chem.soc.,2006,128,12394-12395。noorduin,w.,grinthal,a.,mahadevan,l.,aizenberg,j.,science,2013,340,832-837。oh,m.,mirkin,c.a.,nature,2005,438,651-654。oh,m.,mirkin,c.a.,angew.chem.,int.ed.2006,45,5492-5494。park,k.h.,jang,k.,son,s.u.,sweigart,d.a.,j.am.chem.soc.,2006,128,8740-8741。pevzner,a.,engel,y.,elnathan,r.,tsukernik,a.,barkay,z.,patolsky,f.,nano lett.,2012,12,7-12。ranft,a.,betzler,s.b.,haase,f.,lotsch,b.v.,crystengcomm,2013,15,9296-9300。rieter,w.j.,taylor,k.m.l.,an,h.,lin,w.,lin,w.,j.am.chem.soc.,2006,128,9024-9025。roberts,r.j.,rowe,r.c.,york,p.,powder technology,1991,65 139-146。rowsell,j.lc.,yaghi,o.m.,angew.chem.,int.ed.2005,44,4670。sader,j.e.,chon,j.w.m.,mulvaney,p.,rev.sci.instrum.,1999,70,3967。schilling,c.i.,plietzsch,o.,nieger,m.,muller,t.,s.,eur.j.org.chem.,2011,1743-1754。seo,j.s.,whang,d.,lee,h.,jun,s.i.,oh,j.,jeon,y.j.,kim,k.,nature,2000,404,982。shi,n.,xie,l.,sun,h.,duan,j.,yin,g.,xub,z.,huang,w.,chem.commun.,2011,47,5055-5057。shirman,t.,lam
è
re,j.f.,shimon,l.j.w.,gupta,t.,martin,j.m.l.,van der boom,m.e.,cryst.growth des.,2008,8,3066-3072。sindoro,m.,yanai,n.,jee,a.y.,granick,s.,acc.chem.res.,2014,47,459-469。smulders,m.m.,riddell,i.a.,browne,c.,nitschke,j.r.,chem.soc.rev.,2013,42,1728-1754。spokoyny,a.m.,kim,d.,sumrein,a.,mirkin,c.a.,chem.soc.rev.,2009,38,1218-1227。stock,n.,biswas,s.,chem.rev.,2011,112,933-969。sun,x.,dong,s.,wang,e.,j.am.chem.soc.,2005,127,13102-13103。tabellion,f.m.,seidel,s.r.,arif,a m.,stang,p.j.,j.am.chem.soc.,2001,123,7740。tabor,d.,j.coll.int.sci.1977,58,2-13。tao,a.r.,habas,s.,yang,p.,small,2008,4,310-325。
thompson,a.m.w.c.,hock,j,mccleverty,j.a.,ward,m.d.,inorg.chim.acta.,1997,256,331-334。tomasik,p.,ratajewicz,z.,newkome,g.r.,strekowski,l.e.,《杂环化合物化学:吡啶金属络合物》(chemistry of heterocyclic compounds:pyridine metal complexes)(第6部分,第14卷).(john wiley&sons,inc.,2008)。tuxen,a.,carenco,s.,chintapalli,m.,chuang,c.h.,escudero,c.,pach,e.,jiang,p.,borondics,f.,beberwyck,b.,alivisatos,a.p.,thornton,g.,pong,w.f.,guo,j.,perez,r.,besenbacher,f.,salmeron,m.,j.am.chem.soc.,2013,135,2273-2278。wang,h.,zeng,y.,ma,j.s.,fu,h.,yao,j.,mikhaleva,a.i.,trofimov,b.a.,chem.commun.,2009,5457-5459。wang,c.,liu,d.,lin,w.,j.am.chem.soc.,2013,135,13222-13234。wei,h.,li,b.,du,y.,dong,s.,wang,e.,chem.mater.,2007,19,2987-2993。whitesides,g.m.,grzybowski,b.,science,2002,295,2418-2421。yaghi,o.m.,obkeeffe,m.,ockwig,n.w.,chae,h.k.,eddaoudi,m.,kim,j.,nature,2003,423,705。zhang,w.,jin,w.,fukushima,t.,ishii,n.,aida,t.,angew.chem,int.ed.,2009,48,4747-4750。zhao,x.,xiao,b.,fletcher,a.j.,thomas,k.m.,bradshaw,d.,rosseinsky,m.j.,science,2004,306,1012。zhao,x.y.,liang,d.d.,liu,s.x.,sun,c.y.,cao,r.g.,gao,c.y.,ren,y.h.,su,z.m.,inorg.chem.,2008,47,7133。zhao,d.,timmons,d.j.,yuan,d.,zhou,h.c.,acc.chem.res.,2011,44,123-133。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。