烟草ntoee1基因及其在调控茎叶夹角和株高中的应用
技术领域
1.本发明属于植物基因工程技术领域,具体涉及一个烟草ntoee1基因在调控茎叶夹角和株高中的应用。
背景技术:
2.茎叶夹角即叶片与主茎之间的夹角,是决定栽培烟草株型的主要因素。叶片与茎秆间角度的大小影响到烟草对光、气的竞争。茎叶夹角小,植株叶片直立向上,冠层截光能力低,群体透光率高,有利于中下部叶片获得更多光照;但是也会使植株的光合面积不大,群体光合效率降低,严重时导致减产;相反,叶柄角度开合过大,又影响行间距,不利用作物的合理密植。
3.烟草作为重要的经济作物,目前对于株高改良的研究还较少。烟草株高与产量、品质等存在相关关系,烟草株高是重要的农艺性状之一。重要的是,烟草获得高产要以合理密植为中心,建立合理的群体结构,其中一个很重要的指标就是株高;株高变大能够改善田间郁闭,增强通风透光情况,提高植物下部叶的光合利用效率,从而改善整株的光合利用率,达到增加产量的效果。株高明显地影响田间群体结构状况和小气候,研究它们对利用和改善田间小气候、提高光能利用率和作物产量有着重要意义。
4.综上,茎叶夹角和植株株高是影响烟株株形的重要影响因素,最终可影响作物产量。烟草叶片作为主要收获的器官,合适的茎叶夹角和合理的株高有助于烟株获得理想株形,提高烟草品质、产量,从而能够帮助烟草生物量和经济系数都达到最高水平。因此,如何有效调控影响株形的因素并将其有效利用,并开展对调控茎叶夹角和株高相关基因的详细研究,对烟草产量、品质的提高具有十分重要的研究理论意义和实用价值。
技术实现要素:
5.本技术目的在于提供一个烟草ntoee1基因,及其所编码烟草ntoee1蛋白、以及其在烟草生长发育、茎叶夹角和株高调控中具有十分重要的应用价值。
6.本发明的技术方案如下:
7.本发明第一方面公开了一个烟草ntoee1基因,所述基因包括3141bp,碱基序列如seq id no.1所示。
8.优选地,所述烟草ntoee1基因的所编码的ntoee1蛋白。
9.优选地,所编码的ntoee1蛋白的氨基酸序列如seq id no.2所示。
10.本发明第二方面公开了所述烟草ntoee1基因在烟草中的应用。
11.优选地,将ntoee1基因进行基因编辑后的植株相比于对照植株叶片夹角变小,株高变矮。
12.优选地,基因编辑后的植株,是通过crispr/cas9介导的基因编辑技术,构建了用于敲除ntoee1基因的crispr/cas9编辑载体,经遗传转化后获得了ntoee1基因发生编辑的烟草植株。
13.优选地,用于敲除所述烟草ntoee1基因的重组crispr/cas9表达载体,使转化植株中烟草ntoee1表达量明显降低或不表达。
14.优选地,将ntoee1基因进行过表达后,过表达植株相比于对照植株,叶片夹角变大,株高变高。
15.本发明烟草ntoee1基因在调控烟草茎叶夹角和株高中的应用包括以下步骤:利用烟草ntoee1基因克隆得到烟草茎叶夹角和株高调控基因;利用基因编辑技术获得烟草ntoee1基因的功能缺失的基因编辑载体而降低烟草茎叶夹角和株高;通过过量表达所述烟草ntoee1基因增大烟草茎叶夹角和株高。
16.作为优选,利用基因克隆得到烟草ntoee1基因包括以下步骤:
17.设计特异克隆烟草ntoee1基因的引物序列如seq id no.3和seq idno.4所示;提取烟草叶片rna并反转录为cdna,以此为模板,利用所述seq id no.3和seq idno.4引物进行第一次pcr扩增;pcr扩增条件为:94℃预变性4min;94℃变性30s,56℃退火30s,72℃延伸40s,共25个循环;再72℃终延伸10min;pcr扩增4℃保存备用,或者直接进行电泳检测分析。将上述第二次pcr扩增的产物片段切胶回收后,胶回收目的片段4μl,peasy-t1 vector 1μl,solutioni5μl;16℃反应30min后取连接产物转化dh5α感受态细胞,37℃培养18-20h,对菌板进行菌落pcr,筛选阳性克隆,鉴定获得烟草ntoee1基因,具有如seq id no.1所示序列。
18.作为优选,利用基因编辑技术获得烟草ntoee1基因的功能缺失的基因编辑载体包括以下步骤:选择ntoee1基因中较特异的23nt核苷酸序列(seq id no.5)为crispr/cas9的引导序列,设计敲除引物序列oee1-k-f和oee1-k-r如下:oee1-k-f:5
’‑
gattgcatatggttctgtccgatgg-3’(seq id no.6),oee1-k-r:5
’‑
aaacccatcggacagaaccatatgc-3’(seq id no.7);利用上述引物序列,获得靶位点的dna双链(退火);将所获得靶位点的dna双链(退火产物)与bsai酶切后的crispr/cas9载体进行连接,并转化、筛选、pcr扩增检测,pcr阳性克隆送测序公司进行测序确认,最后得到crispr/cas9-ntoee1编辑载体。利用上一步所构建的crispr/cas9-ntoee1编辑载体质粒,以红花大金元为例,进行遗传转化和组培,以获得ntoee1基因发生敲除编辑的植株。
19.作为优选,通过过量表达烟草ntoee1基因增大茎叶夹角和株高:基因的组织表达模式与其基因功能之间存在着密切的关系,发明人分析了烟草ntoee1基因的组织表达模式情况,结果发现,烟草ntoee1在叶片中表达量最高,在根中表达量最低。利用全基因组表达谱数据,进一步调查了烟草ntoee1组织表达模式,分析发现,烟草ntoee1同样在叶片中具有很高的表达量,在根中表达量最低。这与定量pcr数据结果相一致。由于叶片是光合作用最主要的场所之一,烟草ntoee1在叶片中的高表达模式很可能与其参与的光合作用过程密切相关。通过开展ntoee1基因表达分析,发明人对烟草ntoee1基因在烟草生长中的作用有了初步了解。
20.与现有技术相比,本发明的有益效果在于有效调控烟草的茎叶夹角和株高,有效改善其农艺性状,有助于提高烟草的产量和品质。
附图说明
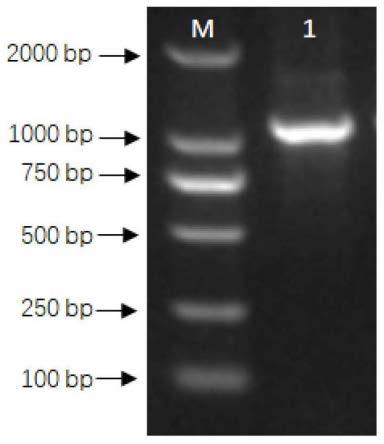
21.图1为本发明实施例所提供的ntoee1基因克隆电泳图谱。
22.图2为ntoee1基因在不同组织中的表达特征;图2a为基于q-pcr不同组织表达模式分析(根中相对表达值为1);图2b为基因芯片表达谱数据的不同组织表达模式分析。
23.图3为未发生基因编辑烟草植株对照样(左边两株)与基因编辑烟草植株(右边两株)整体表型观察。
24.图4为未发生基因编辑烟草植株对照样(左)与基因编辑烟草植株(右)叶夹角的表型观察。
25.图5为ntoee1在过表达植株中表达量分析。
26.图6为对照植株(左)与过表达阳性植株(右)整体表型观察。
27.图7为对照植株(左)与过表达阳性植株(右)叶夹角的表型观察。
具体实施方式
28.下面结合实施例对本技术做进一步的解释说明。
29.在介绍具体实施例前,就下述实施例中涉及部分生物材料、实验试剂、实验设备等情况简要介绍如下。
30.生物材料:
31.烟草品种:红花大金元,一种商品化烟草,实施例中所采用种子由国家烟草基因研究中心保存提供。
32.载体:peasy-t1 simple载体,购自北京全式金生物技术有限公司。
33.crispr/cas9载体由西南大学家蚕基因组生物学国家重点实验室友情提供。
34.菌株:
35.trans1-t1化学感受态细胞,购自北京全式金生物技术有限公司;
36.lba4404农杆菌菌株,生物实验中常用菌株,可公开获得;
37.引物的合成和dna测序由北京六合华大基因科技股份有限公司提供完成;
38.实验试剂:
39.rna提取试剂盒,superpure plant polyrna kit;
40.荧光定量pcr酶(sybr qpcr kit),购自郑州安赛生物科技有限公司;
41.反转录试剂盒、t4连接酶,购自宝生物工程(大连)有限公司;
42.限制性内切酶bsai,购自neb公司;
43.dna扩增酶,购自北京全式金生物技术有限公司;
44.植物基因组提取试剂盒、dna纯化试剂盒购自qiagen公司。
45.实验设备:
46.pcr合成仪tprofessional thermocycler,biometra公司;
47.定量pcr仪cfx96,bio-rad公司;
48.紫外凝胶成像系统biospectrum,uvp公司。
49.实施例1
50.本实施例主要就ntoee1基因的克隆获得过程简要介绍如下。
51.(1)制备cdna作为克隆模板
52.取旺长期烟草(红花大金元)的茎100mg作为样品,在液氮中充分研磨,参照rna提取试剂盒说明书提取总rna,然后反转录为cdna备用;
53.(2)设计引物,进行pcr扩增
54.设计用于扩增烟草ntoee1的基因引物序列如下:
55.ntoee1-f:5'-atggcagcttctctacaagcagctg-3'(seq id no.3),
56.ntoee1-r:5'-ttacgcatcctgccgcggttccacc-3'(seq id no.4);
57.以步骤(1)中所制备cdna为模板,利用上述引物进行pcr扩增,pcr扩增条件为:94℃预变性3min,94℃变性20s,58℃退火20s,72℃延伸30s,35个循环,循环结束后72℃ 10min彻底延伸;pcr扩增4℃保存备用,或者直接进行电泳检测分析。
58.参照胶回收试剂盒说明书纯化pcr扩增后产物,纯化产物再连接至peasy-t1载体上,连接体系如下:
59.dna扩增产物,6μl;
60.peasy-t1载体,1μl;
61.混匀后,25℃连接25min。
62.将连接产物转化至大肠杆菌感受态细胞中,具体转化过程简要介绍如下:
63.从-80℃冰箱中取出感受态细胞,置于冰上使其溶解,加连接产物于50μl的trans1-t1感受态细胞中,轻弹混匀,冰浴30min;
64.42℃水浴中热激30s,立即置于冰上2min;加入250μl平衡至室温的lb(不含抗生素),37℃摇荡培养1h;
65.取8μl的500mm的iptg、40μl的20mg/ml的x-gal,混合后均匀地涂布到lb固体平板(含60μg/μl氨苄青霉素)平板上,倒置培养皿,37℃培养过夜。
66.挑取白斑扩增培养后,提取各质粒dna,通过质粒pcr扩增对重组质粒进行了鉴定,将对应的阳性克隆送样测序,获得ntoee1基因序列。
67.测序分析结果表明,ntoee1基因编码区长度为3141bp个核苷酸,具体如seq id no.1所示;对该基因分析后可知,其所编码的ntoee1蛋白的氨基酸序列如seq id no.2所示。图1为本实施例所提供的ntoee1基因克隆电泳图谱;由图1可知,pcr产物经琼脂糖凝胶电泳检测,获得了1000bp左右大小的pcr条带,测序结果显示,烟草ntoee1基因的蛋白质编码区(cds)全长为1083bp。
68.实施例2
69.利用实施例1中所获得烟草ntoee1基因,进一步构建了crispr/cas9载体,以及利用叶盘法转化获得基因编辑植株。
70.选择ntoee1基因中较特异的23nt核苷酸序列(seq id no.5)为crispr/cas9的引导序列,设计敲除引物序列oee1-k-f和oee1-k-r如下:
71.oee1-k-f:5
’‑
gattgcatatggttctgtccgatgg-3’(seq id no.6),
72.oee1-k-r:5
’‑
aaacccatcggacagaaccatatgc-3’(seq id no.7);
73.利用上述引物序列,获得靶位点的dna双链(退火);将所获得靶位点的dna双链(退火产物)与bsai酶切后的crispr/cas9载体进行连接,并转化、筛选、pcr扩增检测,pcr阳性克隆送测序公司进行测序确认,最后得到crispr/cas9-ntoee1编辑载体。
74.利用上一步所构建的crispr/cas9-ntoee1编辑载体质粒,以红花大金元为例,进行遗传转化和组培,以获得烟草开花时间相关的基因ntoee1发生敲除编辑的植株,相关实验过程简要介绍如下。
75.将烟草种子表面消毒后点种至ms培养基上,待长到4片子叶(15-20d),移入含ms固体培养基的培养瓶中,于25
±
1℃、光照强度30-50μmol/(m2
·
s),光照时间为16h/d条件继续培养35-40d,备用。
76.取出-80℃保存的lba4404电转化感受态农杆菌细胞,置于冰上冻融。待感受态刚刚解冻时,加入crispr/cas9-ntoee1编辑载体质粒的2μl,混匀,置于冰上。后将混匀的感受态转移至预冷的电转杯中,将电转杯置于电转仪中进行转化,转化完成后加入1ml的yeb液体培养基与转化液进行混合,后置于摇床28℃,200rpm培养1.5-2h。8000rpm离心菌体弃掉上清培养基,然后用200μl的yeb液体培养基悬浮菌体,涂于含50mg/l利福平、50mg/l链霉素和50mg/l卡那霉素的yeb固体培养基上28℃倒置黑暗培养2-3d。
77.在超净工作台中制作烟草叶盘成边长为1cm的方形叶盘,用ms液体制备含有crispr/cas9-ntoee1编辑载体的农杆菌菌落成悬浮菌液(od600=0.6-0.8)。利用悬浮农杆菌菌液浸泡侵染烟草叶盘10min。之后将叶盘置于含2.0mg/l naa 0.5mg/l 6-ba的ms固体培养基上,28℃,黑暗,共培养3d。之后进行继代培养,放置于含2.0mg/l naa 0.5mg/l 6-ba 250mg/l cb 50mg/l kan的ms固体培养基上,培养条件为:28℃光照培养16h/d,光照强度30-50μmol/(m2
·
s),25℃黑暗培养8h/d,培养45-60d,直至分化芽形成,每7-10d更换一次分化培养培养基,更换3-4次;培养至分化芽形成;将已有分化芽形成的愈伤组织切下,置于含有500mg/l羧苄青霉素与50mg/l卡那霉素的ms培养基上进行培养,待愈伤组织上分化芽培养长至2-4cm高,培养条件与分化培养条件一致,培养8-14d;再生植株生根培养,将分化芽切下,插入含有500mg/l羧苄青霉素与50mg/l卡那霉素的ms培养基上进行生根培养,培养条件与分化培养条件一致,培养20-30d,再生移栽至花盆后进行培养,后进行转化植株叶片取样,送华大基因进行分子检测,确定获得ntoee1基因编辑植株,之后进行收种获得t0代编辑植株种子。t0代种子按23倍进行自交纯合扩繁,待植株长到5-6片叶时,单株的叶片取样,送华大基因进行分子检测,确定获得ntoee1基因发生纯合编辑的植株,之后进行收种获得ntoee1基因纯合编辑的t1代种子。
78.本发明所述的烟草ntoee1基因的应用为在烟草植株体内降低所述ntoee1基因的表达,基因编辑植株相比于对照叶片夹角明显变小,株高显著变矮,从而可以调控烟草株型。现有技术领域内常用的降低基因表达或者基因沉默的方法均适用于本发明。
79.图2为ntoee1基因在不同组织中的表达特征;图2a为基于q-pcr不同组织表达模式分析(根中相对表达值为1);图2b为基因芯片表达谱数据的不同组织表达模式分析。由图2可以看出烟草ntoee1在叶片中表达量最高,在根中表达量最低。另外,烟草ntoee1在叶脉、花瓣、花萼中具有较高的表达量。利用全基因组表达谱数据,进一步调查了烟草ntoee1组织表达模式,分析发现,烟草ntoee1同样在叶片中具有很高的表达量,在根中表达量最低。这与定量pcr数据结果相一致。
80.实施例3
81.利用实施例2中分子检测确定为ntoee1基因纯合敲除的植株,通过整体观察表型分析,获得基因编辑材料与野生型间的差别,图3为未发生基因编辑烟草植株对照样(左边两株)与基因编辑烟草植株(右边两株)整体表型观察;图4为未发生基因编辑烟草植株对照样(左)与基因编辑烟草植株(右)叶夹角的表型观察。由图3和图4可以看出,ntoee1基因编辑烟草植株的叶夹角和株高都小于未编辑烟草植株。
82.实施例4
83.利用实施例1中所获得烟草ntoee1基因,本发明进一步构建了过表达载体,根据ntoee1基因编码序列,设计过表达载体构建引物:
84.oe-ntoee1-f:5'-cgggatcccgatggcagcttctctacaagcagctg-3'(seq id no.8);
85.oe-ntoee1-r:5'-aaaagtacttttttacgcatcctgccgcggttccacc-3'(seq id no.9)。以peasy-t1-ntoee1为模板进行pcr扩增,扩增产进行pcr切胶回收。
86.a.提取pbi121质粒,用限制性bamhi和scai内切酶分别双酶切oe-ntoee1引物的扩增产物和pbi121质粒。酶切体系如下:
[0087][0088]
酶切条件:37℃,3h。胶回收和纯化目的片段(方法同上),-20℃保存备用。
[0089]
b.载体和目的片段连接
[0090]
将回收的载体和目的片段用t4连接酶连接,反应体系如下:
[0091][0092]
连接条件:16℃过夜连接
[0093]
c.连接产物的转化和阳性克隆质粒的筛选
[0094]
农杆菌感受态的制备与表达载体转化
[0095]
农杆菌感受态的制备
[0096]
a.挑取农杆菌lba4404的单菌落,接种于5ml含50mg/l链霉素和利福平的yeb液体培养基中,200rpm,28℃振荡培养过夜;
[0097]
b.取2ml菌液接种于100ml含50mg/l链霉素和利福平的yeb液体培养基中扩大培养,28℃,200rpm振荡培养至od600=0.6;
[0098]
c.将菌液转入50ml离心管中,冰浴30min,5000rpm,4℃离心10min,收集菌体,弃上清液;
[0099]
d.加入30ml预冷的10%甘油溶液,悬浮细菌,5000rpm,4℃离心10min,收集菌体,弃上清液;
[0100]
e.加入20ml 10%甘油溶液,重新悬浮菌体,5000rpm,4℃离心10min,收集菌体,弃上清液;
[0101]
f.加入2ml 10%甘油溶液,轻轻悬浮菌体,200μl每管分装于预冷的1.5ml离心管
中;
[0102]
g.液氮冷冻1分钟,-80℃保存。
[0103]
ntoee1过表达载体遗传转化与阳性植株鉴定
[0104]
1)叶盘法转化烟草
[0105]
a.取烟草无菌苗叶片,用打孔器将叶片处理成直径0.5cm大小的叶盘,叶背朝下,在ms固体培养基上预培养3天;
[0106]
b.农杆菌的培养:挑取单菌落,在2ml含有rif(100μg/ml)、str(50μg/ml)和kan(50μg/ml)的yeb培养基过夜培养;
[0107]
c.取1ml菌液,加入到50ml含相同抗性的yeb培养基中,28℃,200rmp培养至od达到0.6左右,4000rpm离心5min,用20ml的ms液体培养基悬浮菌体;
[0108]
d.农杆菌侵染叶片:将预培养的叶片放到菌液中,侵染10分钟,用无菌的滤纸吸干叶片多余的菌液,在ms 6-ba(2mg/l) naa(0.5mg/l)的固体培养基上暗培养3天;
[0109]
e.用含有cef(400mg/l)的无菌水清洗叶盘,无菌滤纸吸去多余的液体,将叶再盘转到含有6-ba(2mg/l)、naa(0.5mg/l)、cef(200mg/l)和kan(50mg/l)的ms固体筛选培养基上,28℃光照培养;
[0110]
f.待不定芽长到0.5cm时,转移到含cef(200mg/l)和kan(50mg/l)的ms固体培养基上生根,得到t0代转基因株系。
[0111]
2)阳性植株鉴定
[0112]
按照基因组提前试剂盒提前各转基因植株的dna,设计可以扩增载体序列(35s)和-ntoee1部分序列特异性引物。扩增程序为:94℃预变性3min,94℃变性40s,60℃退火40s,72℃延伸40s,30个循环,循环结束后72℃ 10min彻底延伸。通过琼脂糖凝胶电泳检测是否扩增到目的条带,以野生型红花大金元的dna为模板作为pcr扩增的阴性对照。
[0113]
ntoee1过表达植株基因表达分析
[0114]
利用rna提取试剂盒提取各阳性植株rna、反转录后,利用实时荧光定量pcr分析过表达植株中烟草ntoee1基因表达水平,以野生型红花大金元cdna模板作为qpcr扩增对照,如图5所示,ntoee1基因在转基因材料中表达明显升高,表明已获得的阳性材料是可用的。
[0115]
实施例5
[0116]
利用实施例4中确定为ntoee1基因过量表达的植株,通过整体观察表型分析,获得基因编辑材料与野生型间的差别。图6为对照植株(左)与过表达阳性植株(右)整体表型观察;图7为对照植株(左)与过表达阳性植株(右)叶夹角的表型观察。由图6和图7可以看出,ntoee1过表达阳性植株的叶夹角和株高都大于未进行过表达的对照烟草植株。
[0117]
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
[0118]
[0119]
[0120]
[0121]
[0122]
[0123]
[0124]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
