1.本发明属于化合物提取技术领域,涉及一种抑制黄嘌呤氧化酶的沙棘提取物的制备方法及应用;具体涉及一种具有抑制黄嘌呤氧化酶活性的沙棘提取物及其制备方法、应用。
背景技术:
2.现有技术中,黄嘌呤氧化酶(xo)是存在于包括人类在内的各种脊椎动物中的胞质酶,属于钼黄酮蛋白家族,其广泛存在于肾脏、肺、心脏和血管内皮中。黄嘌呤氧化酶是包括人类在内的一些物种中控制尿酸产生的关键酶,控制着人体内嘌呤代谢中次黄嘌呤转化为黄嘌呤和黄嘌呤转化为尿酸的两部关键反应,并且其在肝脏和肠道中活性最好。黄嘌呤氧化酶的活性过度活跃,则会引发体内尿酸含量升高,从而导致高尿酸血症,痛风等疾病。
3.沙棘果(hippophae fructus,别名:达尔,沙枣),属于胡颓子科沙棘属植物沙棘的成熟的种子,主要分布在华北、西北、西南等地。沙棘是中国一种传统中药,自古便被用于温养脾气,开胃消食,活血祛瘀;也能用于治疗止咳祛痰,胸痹心痛,跌打损伤,妇女月经不调等症状。在药理学研究方面,沙棘果具有降低胆固醇,治疗心绞痛、慢性气管炎、胃和十二指肠溃疡、消化不良等疾病,其外还具有防治冠状动脉粥样硬化性心脏病的作用。但是,沙棘果在抗高尿酸血症方面研究较少。
技术实现要素:
4.针对上述问题,本发明提供了一种具有黄嘌呤氧化酶抑制活性的沙棘提取物和7种天然化合物。
5.本发明还提供了一种上述沙棘果提取物的制备方法。
6.本发明还提供了一种上述沙棘果提取物的应用。
7.本发明的技术方案是:本发明所述的一种抑制黄嘌呤氧化酶的沙棘提取物的制备方法,所述沙棘提取物具有下述通式(i)的结构:
8.[0009][0010]
进一步的,其具体制备步骤如下:
[0011]
步骤(1)、干燥沙棘果利用80%乙醇提取,抽滤,收集滤液,滤液减压浓缩,制得沙棘乙醇提取物;
[0012]
在制得的沙棘乙醇提取物中加入5l纯净水进行溶解,再分别利用石油醚、乙酸乙酯和正丁醇依次萃取,分别制得石油醚萃取物、乙酸乙酯萃取物及正丁醇萃取物;
[0013]
步骤(2)、将乙酸乙酯萃取物使用硅胶柱进行分离,并使用二氯甲烷/甲醇体系梯度进行洗脱,从而产生七个组分,即:fr 1、fr 2、fr 3、fr 4、fr 5、fr 6及fr 7;
[0014]
步骤(3)、将组分中产生的fr 5使用硅胶柱进行分离,并使用石油醚/乙酸乙酯体系梯度洗脱,从而获得八个组分,即fr 5a、fr 5b、fr 5c、fr 5d、fr 5e、fr 5f、fr 5g及fr 5h;
[0015]
其中,在获得的八个组分中,fr 5g先经过sephadex lh-20富集,后反相ods粗分除杂,最后和半制备型hplc纯化,从而得到化合物1;
[0016]
步骤(4)、将组分中产生的fr 6使用硅胶柱进行分离,并使用石油醚/乙酸乙酯体系梯度洗脱,从而得到七个组分,即fr 6a、fr 6b、fr 6c、fr 6d、fr 6e、fr 6f及fr 6g;
[0017]
其中,在获得的七个组分中,fr 6c在thf/ch2cl2混合体系中重结晶,从而得到化合物2;
[0018]
fr 6d经过sephadex lh-20富集除杂后,在thf/ch2cl2混合体系重结晶,从而得到化合物3;
[0019]
fr 6e经过sephadex lh-20富集除杂后,用半制备型hplc分离,从而得到化合物4;
[0020]
步骤(5)、将组分中产生的fr 7使用硅胶柱进行分离,并使用石油醚/乙酸乙酯体系梯度洗脱,从而获得七个组分,即:fr 7a、fr 7b、fr 7c、fr 7d、fr 7e、fr 7f、fr 7g及fr7h;
[0021]
其中,在获得的七个组分中,fr 7c硅胶柱进行分离,并使用二氯甲烷/甲醇体系等度洗脱,从而得到:fr 7c.1和fr 7c.2;
[0022]
fr 7c.1和fr 7c.2再经过sephadex lh-20分离后,在二氯甲烷/甲醇体系中重结晶,从而分别得到化合物5和化合物6。
[0023]
最后,将用试管接到的经过硅胶柱的石油醚/乙酸乙酯洗脱剂静置,待溶剂挥发后,结晶得到化合物7。
[0024]
进一步的,在步骤(1)中,所述沙棘乙醇提取物的具体提取过程为:将10kg沙棘在80℃下用3
×
20l的80%乙醇提取2h,提取液过滤后减压浓缩,从而得到无醇味的沙棘乙醇提取物。
[0025]
进一步的,在步骤(2)中,所述硅胶柱为100-200目;
[0026]
在所述逐步洗脱中,ch2cl2/meoh的体积比为:100/1、80/1、50/1、20/1、10/、8/10、6/1、4/1、2和1/1。
[0027]
进一步的,在步骤(3)中,所述分离fr 5的硅胶柱所用硅胶为200-300目;
[0028]
所述石油醚/乙酸乙酯体系的体积比为:15/1-2/1;
[0029]
在所述sephadex lh-20中,流动相为meoh/ch2cl2=1/1;
[0030]
所述反相ods梯度洗脱所用流动相为:ma/h2o,其体积比分别为:10/90、30/70、50/50、80/20;
[0031]
所述化合物1的半制备型hplc的条件为:λ=210-385nm,流速为2.5ml/min;流动相:ma:h2o(0.3%甲酸)=63:37(v/v);出峰时间为22.3min。
[0032]
进一步的,在步骤(4)中,所述硅胶柱所用硅胶为200-300目,流动相为体积比为:3/1-1.5/1的石油醚/乙酸乙酯;
[0033]
所述sephadex lh-20中,流动相为:meoh;
[0034]
所述化合物2重结晶的thf/ch2cl2体系体积比为:1/42;
[0035]
所述化合物3重结晶的thf/ch2cl2体系体积比为:1/50;
[0036]
所述化合物4的半制备hplc的条件为:λ=210-385nm,流速为2.5ml/min,流动相为ma:h2o=35:65,0.3%甲酸,出峰时间为23.0min。
[0037]
进一步的,在步骤(5)中,所述分离fr 7的硅胶柱所用硅胶为200-300目,石油醚/乙酸乙酯体系的体积比为2/1-1/4;
[0038]
所述分离fr 7c的硅胶柱所用硅胶为200-300目,二氯甲烷/甲醇体系的体积比为:15/1;
[0039]
所述sephadex lh-20中,流动相为meoh;
[0040]
所述化合物5重结晶的ch2cl2/ma体系体积比为:4.5/1;
[0041]
所述化合物6的重结晶的ch2cl2/ma体系体积比为:2.5/1;
[0042]
在制备化合物7中,所述石油醚/乙酸乙酯的摩尔比是:1:1。
[0043]
进一步的,所述制备方法制备的一种抑制黄嘌呤氧化酶的沙棘提取物在制备抑制黄嘌呤氧化酶药物中的应用。
[0044]
本发明的沙棘提取物和化合物具有良好的黄嘌呤氧化酶抑制活性。以高效液相色谱(hplc)为检测手段,实验对沙棘提取物和将沙棘乙酸乙酯部分硅胶分离得到的fr 1-fr 7部分进行黄嘌呤氧化酶活性检测,结果发现沙棘提取物和fr5-fr 6均具有显著活性。因此,沙棘提取物表现出优异的黄嘌呤氧化酶抑制活性。表明沙棘在抗高尿酸血症方面具有巨大潜力。
[0045]
本发明的有益效果是:本发明的特点是:1、本发明通过优化反应条件,最大程度的提取了沙棘中的有效成分,提取得到的沙棘提取物表现出优异的黄嘌呤氧化酶抑制活性,在抗高尿酸血症方面具有巨大潜力;2、本发明对沙棘的抗黄嘌呤氧化酶的主要活性成分及深入系统的研究,许多具有显著活性的化合物,不仅丰富了沙棘植物的化合物研究的范畴,
也为沙棘这种药食两用植物进一步功能食品方面的应用提供了一定的理论指导,为该植物的深入开发奠定了基础。
附图说明
[0046]
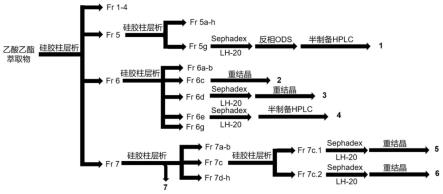
图1是本发明实施例1沙棘乙酸乙酯部分的化合物分离过程示意图;
[0047]
图2是本发明化合物1的氢、碳谱图,其中(a)是氢谱图,(b)是碳谱图;
[0048]
图3是本发明中化合物2的氢谱图;
[0049]
图4是本发明中化合物3的氢谱图;
[0050]
图5是本发明化合物4的氢、碳谱图,其中(a)是氢谱图,(b)是碳谱图;
[0051]
图6是本发明化合物5的氢、碳谱图,其中(a)是氢谱图,(b)是碳谱图;
[0052]
图7是本发明化合物6的氢、碳谱图,其中(a)是氢谱图,(b)是碳谱图;
[0053]
图8是本发明化合物7的氢、碳谱图,其中(a)是氢谱图,(b)是碳谱图。
具体实施方式
[0054]
为了更清楚地说明本发明的技术方案,下面结合附图对本发明的技术方案做进一步的详细说明:
[0055]
本发明提供了一种具有黄嘌呤氧化酶抑制活性的沙棘提取物的制备方法具体制备步骤如下:
[0056]
(1)、干燥沙棘果利用80%乙醇提取,抽滤,收集滤液,滤液减压浓缩,得沙棘乙醇提取物提物;提取物加入5l蒸馏水稀释,分别利用石油醚、乙酸乙酯和正丁醇依次萃取,分别得到石油醚萃取部分(hf-pf),乙酸乙酯萃取部分(hf-ef)和正丁醇萃取部分(hf-bf);
[0057]
(2)、沙棘乙酸乙酯萃取部分通过硅胶柱分离,并用ch2cl2/meoh逐步洗脱产生六个组分fr 1-fr7;
[0058]
(3)、使用石油醚/乙酸乙酯将fr5于硅胶柱分离获得八个组分fr5a到fr5h;其中fr5g经过sephadexlh-20纯化后,利用反相ods梯度洗脱,再用半制备型hplc得到化合物1;
[0059]
(4)、fr 6于硅胶柱用石油醚/乙酸乙酯分离获得七个组分fr6a到fr6g;
[0060]
其中化合物2从fr6c中在thf/ch2cl2体系中重结晶得到,fr6d经过sephadex lh-20分离后,再用thf/ch2cl2体系重结晶得到化合物3;fr 6e经过sephadex lh-20分离后,再用半制备型hplc得到化合物4;
[0061]
(5)、fr 7经过石油醚/乙酸乙酯梯度洗脱后,得到fr 7a到fr7h,其中化合物7在石油醚/乙酸乙酯洗脱下来静置后结晶;fr 7c再度使用硅胶等度分离和sephadex lh-20分离,在dc/ma体系中重结晶得到化合物5和化合物6。
[0062]
进一步的,步骤(1)中,具体过程为:将10kg沙棘在80℃下用3
×
20l的80%乙醇提取2h,提取液过滤后减压浓缩,得到无醇味的提取物。
[0063]
进一步的,步骤(2)中,所述硅胶柱为100-200目;所述逐步洗脱中,ch2cl2/meoh的体积比为:100/1
→
50/1
→
20/1
→
10/
→
8/10
→
6/1
→
4/1
→
2/1
→
1/1
→
ma。
[0064]
进一步的,步骤(3)中,所述石油醚/乙酸乙酯的体积比为15/1-2/1;所述分离fr 5及fr 5g的硅胶柱为200-300目;所述sephadex lh-20中,流动相为meoh/ch2cl2=1/1;所述ods反相分离的梯度洗脱流动相为ma/h2o,体积比分别为10/90,30/70,50/50,80/20;所述
化合物1的半制备hplc的条件为:λ=210-385nm,2.5ml/min;流动相为:为ma:h2o=63:37,0.3%甲酸,出峰时间为22.3min。
[0065]
进一步的,步骤(4)中,所述硅胶柱所用硅胶为200-300目,流动相为体积比为3/1-1.5/1的石油醚/乙酸乙酯;所述sephadex lh-20中,流动相为meoh;所述化合物2重结晶的thf/ch2cl2体系体积比为1/42;所述化合物3重结晶的thf/ch2cl2体系体积比为1/50;所述化合物4的半制备hplc的条件为λ=210-385nm,流速为2.5ml/min,流动相为ma:h2o=35:65,0.3%甲酸,出峰时间为23.0min。
[0066]
进一步的,步骤(5)中,所述fr 7的硅胶柱分离所用硅胶为200-300目,流动相为体积比为2/1-1/4的石油醚/乙酸乙酯;fr 7c的硅胶柱分离所用硅胶为200-300目,流动相为体积比为15/1的二氯甲烷/甲醇;所述sephadex lh-20中,流动相为meoh;所述化合物5重结晶的ch2cl2/ma体系体积比为4.5:1;所述化合物6的重结晶的ch2cl2/ma体系体积比为2.5:1。所述化合物7在石油醚/乙酸乙酯(1:1)洗脱下来静置后结晶。
[0067]
本发明还提供了一种利用上述制备方法制备得到的沙棘提取物的主要活性成分,结构式如下:
[0068][0069]
本发明还提供了一种上述沙棘提取物和分离得到的化合物在制备抑制黄嘌呤氧化酶药物中的应用。
[0070]
实施例:
[0071]
(1)、干燥沙棘果(10kg)在80℃下用3
×
20l的80%乙醇提取2h,提取液过滤后减压浓缩,得到乙醇提取物;将提取物用蒸馏水稀释(5l)后,依次用石油醚(pe,3
×
4.5l)、乙酸乙酯(ea,3
×
4.5l)和正丁醇(n-bu,3
×
4.5l)萃取,分别得到石油醚萃取物部分(pf)150g,乙酸乙酯萃取物部分(ef)80g和正丁醇萃取物部分(bf)1022g;
[0072]
(2)、沙棘乙酸乙酯提取物通过硅胶柱(100-200目)分离,并用ch2cl2/meoh逐步洗
脱(100/1
→
50/1
→
20/1
→
10/
→
8/10
→
6/1
→
4/1
→
2/1
→
1/1
→
ma)产生七个组分(fr 1-fr 7);分别取2mg三种萃取物,用少量dmso促溶后,用缓冲液稀释至10ml,得到200μg
·
ml-1
的实验溶液,对七个组分进行黄嘌呤氧化酶活性检测,结果如图表1所示,fr5-fr7活性较高,因此对其进行分离。
[0073]
表1
[0074][0075]
(3)、使用石油醚/乙酸乙酯(15/1-2/1)将fr 5于硅胶柱(200-300目)分离获得八个组分fr 5a到fr 5h;其中fr 5g经过sephadex lh-20(ma)分离,反相ods(ma/h2o=10/90,30/70,50/50,80/20)梯度洗脱后,再用半制备型hplc得到化合物1(λ=210-385nm,流速为2.5ml/min,流动相为ma:h2o=63:37,0.3%甲酸,出峰时间为22.3min)和化合物2(λ=210-385nm,流速为2.5ml/min,流动相为ma:h2o=50:50,0.3%甲酸,出峰时间为49.5min);fr 6于硅胶柱用石油醚/乙酸乙酯(3/1-1.5/1)分离获得七个组分fr 6a到fr 6g;其中化合物3从fr 6b中在thf/ch2cl2体系(1/42)中重结晶得到,fr 6d经过sephadex lh-20(ma)分离后,再用thf/ch2cl2体系(1/50)重结晶得到化合物4;fr 6e经过sephadex lh-20(ma)分离后,再用半制备型hplc得到化合物5(λ=210-385nm,流速为2.5ml/min,流动相为ma:h2o=35:65,0.3%甲酸,出峰时间为23.0min);fr 7经过石油醚/乙酸乙酯(2/1-1/4)梯度洗脱后,得到fr 7a到fr 7h,其中化合物7在石油醚/乙酸乙酯(1:1)洗脱下来静置后结晶;fr 7c再度使用硅胶等度分离和sephadex lh-20(ma)分离,在dc/ma体系中重结晶得到化合物5(4.5/1)和化合物6(2.5/1);(具体分离图如图1所示)。
[0076]
(4)、化合物1-7鉴定过程:
[0077]
化合物1:白色固体,易溶于甲醇;ei-ms m/z:253.0932[m h]
,结合核磁碳信号,分子式确定为c
15h12
n2o2,不饱和度为11;化合物1为5-hydroxyaspastipuline,谱图如2所示。
[0078]
化合物2:黄色色固体,不溶于二氯甲烷,溶于甲醇;ei-ms m/z:317.0617[m h]
,结合核磁碳信号,分子式确定为c
16h12
o7,不饱和度为11;化合物2为3-甲氧基槲皮素(3-o-methylquercetin),谱图如3所示。
[0079]
化合物3:黄色固体,不溶于二氯甲烷,溶于甲醇;ei-ms m/z:303.0460[m h]
,结合核磁碳信号,分子式确定为c
15h10
o7,不饱和度为11;化合物3为槲皮素(quercetin),谱图如4所示。
[0080]
化合物4:黄色固体,不溶于二氯甲烷,易溶于甲醇;ei-ms m/z:305.0617[m h]
,结合核磁碳信号,分子式确定为c
15h12
o7,不饱和度为11;化合物4为花旗松素(taxifolin),谱图如5所示。
[0081]
化合物5:白色固体,易溶于甲醇;ei-ms m/z:190.0459[m h]
,结合核磁碳信号,分子式确定为c
10
h7no3,不饱和度为8;化合物5为1-oxo-2h-isoquinoline-4-carboxylic acid,谱图如6所示。
[0082]
化合物6:黄色固体,不溶于二氯甲烷,易溶于甲醇;ei-ms m/z:319.0409[m h]
,结合核磁碳信号,分子式确定为c
15h10
o8,不饱和度为11;化合物6为杨梅素(myricetin),谱
图如7所示。
[0083]
化合物8:黄色固体,不溶于二氯甲烷,溶于甲醇;ei-ms m/z:463.1196[m h]
,结合核磁碳信号,分子式确定为c
22h22o11
,不饱和度为12;化合物8为isorhamnetin7-o-α-l-rhamnoside,谱图如8所示。
[0084]
对从沙棘乙酸乙酯提取物中分离鉴定的化合物7个化合物(1-7)进行黄嘌呤氧化酶抑制活性检测。实验结果如表2所示。
[0085]
表2
[0086][0087]
最后,应当理解的是,本发明中所述实施例仅用以说明本发明实施例的原则;其他的变形也可能属于本发明的范围;因此,作为示例而非限制,本发明实施例的替代配置可视为与本发明的教导一致;相应地,本发明的实施例不限于本发明明确介绍和描述的实施例。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
