1.本发明涉及聚氨酯材料技术领域,尤其涉及一种聚氨酯及制备、超分子聚氨酯弹性体及制备和应用。
背景技术:
2.随着现代科学技术的不断进步与发展,全球对于高性能的结构功能材料的需求逐年增强。对于大多数结构材料来说,强度和韧性是评价材料机械性能的至关重要的指标。聚氨酯材料,作为一类新兴的高分子材料,具有独特的结构特性、可控的物理化学特性和极大的应用潜力。尽管目前聚氨酯(pu) 材料的发展已经达到了很高的水平,但由于韧性(即抗破坏性)和强度通常是相互排斥的,因此在制造高性能pu材料方面仍然存在许多机会和挑战。如今,人们不断追求越来越强、越来越韧的高分子材料,然而在大多数材料中,这两种性能(强度和韧性)往往是相互排斥的。在先进材料的发展中,机械性能(强度和韧性)是评价几乎所有工程结构材料的适用性和耐久性的最基本指标之一。一般来说,传统的优化抗拉强度的策略往往是以牺牲材料的韧性为代价的,因而很难同时满足强度和韧性的要求。截至目前,高分子材料始终存在着强度与韧性难以同时匹配这一问题,制备具备高强度兼高韧性的超分子聚氨酯仍面临着艰巨的挑战。
技术实现要素:
3.本发明的目的在于提供一种聚氨酯及制备、超分子聚氨酯弹性体及制备和应用,所述超分子聚氨酯弹性体具有高强高韧性。
4.为了实现上述发明目的,本发明提供以下技术方案:
5.本发明提供了一种基于四重氢键的超分子聚氨酯,具有式i所示结构:
[0006][0007]
式i;
[0008]
式i中,n=8~27。
[0009]
本发明提供了上述技术方案所述基于四重氢键的超分子聚氨酯的制备方法,包括以下步骤:
[0010]
将聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合,进行预聚反应,得到预聚物;
[0011]
将所述预聚物与t型扩链剂混合,进行扩链反应,固化后,得到基于四重氢键的超分子聚氨酯;
[0012]
所述t型扩链剂具有式ii所示结构:
[0013][0014]
优选的,所述催化剂为二月桂酸二丁基锡;所述聚四氢呋喃二醇、异佛尔酮二异氰酸酯和催化剂的摩尔比为10:20:0.1。
[0015]
优选的,所述预聚反应的温度为80℃,时间为4h。
[0016]
优选的,所述聚四氢呋喃二醇与t型扩链剂的摩尔比为10:(5~10)。
[0017]
优选的,所述扩链反应的温度为80℃,时间为2h。
[0018]
本发明提供了一种超分子聚氨酯弹性体的制备方法,包括以下步骤:
[0019]
将聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合,进行预聚反应,得到预聚物;
[0020]
将所述预聚物与t型扩链剂混合,进行扩链反应后,向所得聚氨酯产物中加入锌盐溶液,进行配位反应,固化后,得到超分子聚氨酯弹性体。
[0021]
优选的,所述锌盐溶液中锌离子与t型扩链剂的摩尔比为(1.67~5):5,所述配位反应的温度为40℃,时间为5h。
[0022]
本发明提供了上述技术方案所述制备方法制备得到的超分子聚氨酯弹性体。
[0023]
本发明提供了上述技术方案所述超分子聚氨酯弹性体在柔性机器人、可穿戴电子设备或自修复薄膜电极中的应用。
[0024]
本发明提供了一种聚氨酯,所述聚氨酯的聚合物链段中的upy基团通过二聚形成四重氢键和通过金属配位作用所形成的锌离子配位键,不仅可以诱导相分离从而形成软硬段结构,还可通过π-π堆积相互作用,在环境温度下形成稳定的微晶,进一步提高聚氨酯材料的机械强度。此外,柔性聚四氢呋喃醚二醇链段上氨基甲酸酯基团之间存在的弱氢键作用,赋予了材料超韧特性。氢键和金属配位键充当牺牲键的作用,在外力下可以发生解离和重构,这一过程需要耗散能量以保护链的完整性。通过有效的能量耗散,抑制应力集中,非共价键还会促进分子链的定向结晶,从而提高强度、韧性,甚至自愈能力。因此,本发明基于多重氢键和金属配位键协同作用,将氢键作用(单重氢键、双重氢键和四重氢键)、金属配位键作用(锌离子和嘧啶酮基团的配位作用)两种超分子相互作用(非共价相互作用)的协同效应整合到聚氨酯骨架中,利用非共价相互作用实现了聚氨酯强度和韧性的协同提升,得到高强韧一体的超分子聚氨酯材料。
[0025]
本发明提供的超分子聚氨酯弹性体中的非共价作用主要包括:四重氢键 (upy-upy)、单重氢键(氨基甲酸酯-氨基甲酸酯)和金属配位键(zn-upy)三种相互作用。因此,基于多重氢键和金属配位键的协同增强机制将为设计高韧性和高强度的聚氨酯弹性体开辟新的可能性,拓宽其在柔性机器人、可穿戴电子设备、自修复薄膜电极等方面的应用潜力和
价值。
附图说明
[0026]
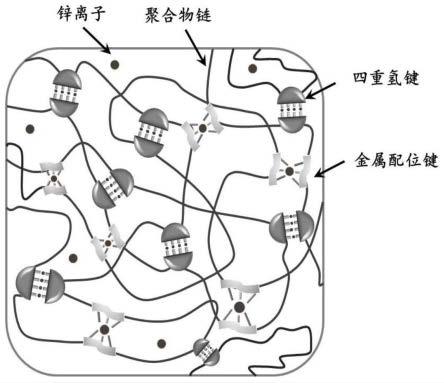
图1为本发明提供的超分子聚氨酯弹性体的网络结构示意图;
[0027]
图2为本发明制备的upy-nco和upy-ampd的红外谱图;
[0028]
图3为实施例1制备的聚氨酯以及实施例2~4制备的超分子聚氨酯弹性体的红外谱图;
[0029]
图4为实施例1和对比例1~2制备的产品的应力-应变曲线图;
[0030]
图5为实施例1~4制备的产品的应力-应变曲线图。
具体实施方式
[0031]
本发明提供了一种基于四重氢键的超分子聚氨酯,具有式i所示结构:
[0032][0033]
式i中,n=8~27。
[0034]
在本发明中,所述n优选为13。
[0035]
本发明提供了上述技术方案所述基于四重氢键的超分子聚氨酯的制备方法,包括以下步骤:
[0036]
将聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合,进行预聚反应,得到预聚物;
[0037]
将所述预聚物与t型扩链剂混合,进行扩链反应,固化后,得到基于四重氢键的超分子聚氨酯;
[0038]
所述t型扩链剂具有式ii所示结构:
[0039][0040]
在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
[0041]
本发明将聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合,进行预聚反应,得到预聚物。
[0042]
在本发明中,所述聚四氢呋喃二醇的分子量优选为600~2000g/mol,更优选为
1000g/mol。
[0043]
在本发明中,所述催化剂为二月桂酸二丁基锡;所述聚四氢呋喃二醇、异佛尔酮二异氰酸酯和催化剂的摩尔比优选为10:20:0.1。
[0044]
在本发明中,所述有机溶剂优选为n,n-二甲基乙酰胺(dmac)或n,n
‑ꢀ
二甲基甲酰胺(dmf)。本发明对所述有机溶剂的具体用量没有特殊的限定,能够将物料充分溶解即可。
[0045]
在本发明中,所述聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合的过程优选包括将将聚四氢呋喃二醇和有机溶剂混合,在110℃油浴条件、n2氛围下搅拌1h,以除去溶剂中的水残留,避免多余水分干扰;待混合液冷却至80℃时,在n2气氛下,加入异佛尔酮二异氰酸酯(ipdi)和二月桂酸二丁基锡。
[0046]
在本发明中,所述预聚反应的温度优选为80℃,时间优选为4h;所述预聚反应优选在搅拌条件下进行;本发明对所述搅拌的过程没有特殊的限定,按照本领域熟知的过程保证反应顺利进行即可。
[0047]
得到预聚物后,本发明优选不进行处理,直接将所述预聚物与t型扩链剂混合,进行扩链反应,固化后,得到基于四重氢键的超分子聚氨酯。在本发明中,所述聚四氢呋喃二醇与t型扩链剂的摩尔比优选为10:(5~10)。
[0048]
在本发明中,所述t型扩链剂优选以溶液的形式使用,所述t型扩链剂的溶液所用溶剂优选为n,n-二甲基乙酰胺(dmac);本发明对所述t型扩链剂的溶液的浓度没有特殊的限定,能够满足其摩尔比要求即可;在本发明的实施例中,所述t型扩链剂的浓度为0.25mmol/ml。
[0049]
在本发明中,所述t型扩链剂具有式ii所示结构:
[0050][0051]
在本发明中,所述t型扩链剂优选按照本领域公知的方法制备,所述t 型扩链剂的制备方法优选包括:
[0052]
将2-氨基-4-羟基-6-甲基嘧啶(mic,5.00g,40mmol)与六亚甲基二异氰酸酯(hdi,40.32g,240mmol)混合在圆底烧瓶中,并在n2气氛、100℃下搅拌 24h,将得到的混合物冷却至室温,并加入过量正戊烷沉淀产物并除去多余的未反应hdi,将所得产物用正戊烷洗涤沉淀3次以上,在50℃下真空干燥12h,得到异氰酸酯封端的2-脲-4[h]-嘧啶酮,记为upy-nco。
[0053]
将所述upy-nco(12.60g,43mmol)、2-氨基-2-甲基-1,3-丙二醇(ampd) (7.03g,66.9mmol)和450ml无水氯仿置于装有冷凝管的圆底烧瓶中,在n2气氛、60℃条件下,回流10h,直至反应完全后,将所得乳白色浑浊溶液真空抽滤,使用大量氯仿洗涤3次,再将所得粉末溶于dmf中,采用离心分离(9000r/min,10min)后取上清液,向上清液中倒入1000ml乙醚进行沉淀,抽滤,真空干燥,得到upy-ampd,即t型扩链剂。
[0054]
在本发明中,所述upy-nco和upy-ampd的制备反应过程为:
[0055]
。
[0056]
本发明优选将所述t型扩链剂的溶液滴加至所述预聚物中;本发明对所述滴加的速率没有特殊的限定,按照本领域熟知的过程进行即可。
[0057]
在本发明中,所述扩链反应的温度优选为80℃,时间优选为2h;所述扩链反应优选在搅拌条件下进行;本发明对所述搅拌的过程没有特殊的限定,按照本领域熟知的过程保证反应顺利进行即可。
[0058]
完成所述扩链反应后,本发明优选将所得产物冷却至室温,滴加聚醚胺 (d230),进行固化,得到聚氨酯,记为spu-upy;所述固化的温度优选为 40℃,时间优选为3h;所述固化优选在搅拌条件下进行,本发明对所述搅拌没有特殊的限定,按照本领域熟知的过程进行即可。
[0059]
在本发明的实施例中,为了制成薄膜样品,完成所述固化后,本发明优选将所得产物倒入四氟模具中,置于真空干燥箱中,在80℃干燥48h,(完全挥发溶剂),得到聚氨酯透明薄膜。
[0060]
在本发明中,所述预聚反应和扩链反应的反应过程为:
[0061]
。
[0062]
本发明提供了一种超分子聚氨酯弹性体的制备方法,包括以下步骤:
[0063]
将聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合,进行预聚反应,得到预聚物;
[0064]
将所述预聚物与t型扩链剂混合,进行扩链反应后,向所得聚氨酯产物中加入锌盐溶液,进行配位反应,固化后,得到超分子聚氨酯弹性体。
[0065]
在本发明中,所述将聚四氢呋喃二醇、异佛尔酮二异氰酸酯、催化剂和有机溶剂混合,进行预聚反应,得到预聚物的过程以及原料配比与上述制备聚氨酯的过程相同,在此不再赘述。
[0066]
得到预聚物后,本发明将所述预聚物与t型扩链剂混合,进行扩链反应后,向所得聚氨酯产物中加入锌盐溶液,进行配位反应,固化后,得到超分子聚氨酯弹性体。
[0067]
在本发明中,将所述预聚物与t型扩链剂混合,进行扩链反应的过程和原料配比与上述制备聚氨酯的过程相同,在此不再赘述。
[0068]
在本发明中,所述锌盐溶液中锌盐优选为氯化锌;所述锌盐溶液所用溶剂优选为dmac;所述锌盐溶液的浓度优选为1mmol/ml。
[0069]
在本发明中,所述锌盐溶液中锌盐与t型扩链剂的摩尔比优选为 (1.67~5):5,更优选为2.5:5,所述配位反应的温度优选为40℃,时间优选为 5h;所述配位反应优选在搅拌条件下进行,本发明对所述搅拌的速率没有特殊的限定,按照本领域熟知的过程保证反应顺利进行即可。
[0070]
在所述配位反应过程中,锌离子与2-脲-4[h]-嘧啶酮基团上的n、o、h 形成配位。
[0071]
完成所述配位反应后,本发明优选将所得产物冷却至室温,滴加聚醚胺 (d230),进行固化;所述固化的温度优选为40℃,时间优选为3h;所述固化优选在搅拌条件下进行,本发明对所述搅拌没有特殊的限定,按照本领域熟知的过程进行即可。
[0072]
完成所述固化后,本发明将所得产物倒入至四氟模具中并在80℃下真空干燥48h,完全除去溶剂,烘干,得到超分子聚氨酯弹性体,记为spu-upy-zn。本发明对所述烘干的过程没有特殊的限定,按照本领域熟知的过程进行即可。
[0073]
本发明提供了上述技术方案所述制备方法制备得到的超分子聚氨酯弹性体,所述超分子聚氨酯弹性体结构示意如式iii所示:
[0074][0075]
本发明所述超分子聚氨酯弹性体存在四重氢键和金属配位键,超分子相互作用之间的键合关系如图1所示,最终所制备的超分子聚氨酯弹性体其聚合物网络结构中包含四重氢键和金属配位键,这两种超分子相互作用。具体的,多重氢键的存在,不仅可以实现断裂后的快速顺序重组,而且可以有效地以较弱的非共价键形式耗散能量,赋予弹性体较好的可拉伸性和弹性。金属配位键作用:由zn
2
离子与upy基团配位组成的zn-upy配位键作为较强的非共价键,有助于形成强大的物理交联网络,从而显著增强弹性体的机械强度。由于优化的四重氢键和金属配位键的协同作用,所得到的超分子聚氨酯弹性体表现出高的拉伸强度、卓越的韧性以及较大的杨氏模量。
[0076]
与传统的化学共价键相比,非共价相互作用的自组装和连接具有动力学和可逆性,可以通过非共价键的断裂和重构去有效地耗散应变能,从而起到硬化网络的作用。一方面,非共价键以牺牲和可逆的方式发挥作用,在结构系统被破坏之前优先断裂,并在外部负载下经历可逆的键断裂和重组,这一过程为材料性能的增强提供了有效的能量耗散。另一方面,非共价键的聚合和重组增加了交联密度致使链的流动性受限,减慢自组装单元的扩散并减少了可用位点寻求键交换的机会,从而阻碍了键的重排和抑制交换反应,有效地避免应力集中在较短的链上、延迟断裂,获得更高的拉伸性和强度。本发明通过结合多种超分子相互作用(氢键和金属配位键两种非共价相互作用) 以协同实现聚氨酯材料机械性能的协同增强,多个氢键和金属配位键的协同效应可以提供很大的灵活性来增强聚氨酯并显著提高其韧性,构建得到集高强度与高韧性为一体的超分子聚氨酯材料。本发明基于分层级的强键和弱键结合的非共价相互作用,构建具有易加工性、可拉伸性、高韧性和坚固性的超分子聚氨酯材料。
[0077]
本发明提供了上述技术方案所述超分子聚氨酯弹性体或上述技术方案所述制备方法制备得到的超分子聚氨酯弹性体在柔性机器人、可穿戴电子设备或自修复薄膜电极中的应用。本发明对所述应用的方法没有特殊的限定,按照本领域熟知的方法应用即可。
[0078]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0079]
以下实施例中,t型扩链剂(upy-ampd)的制备方法为:
[0080]
将2-氨基-4-羟基-6-甲基嘧啶(mic,5.00g,40mmol)与六亚甲基二异氰酸酯(hdi,40.32g,240mmol)混合在圆底烧瓶中,并在n2气氛、100℃下搅拌24h,将得到的混合物冷却至室温,并加入过量正戊烷沉淀产物并除去多余的未反应hdi,将所得产物用正戊烷洗涤沉淀3次以上,在50℃下真空干燥 12h,得到异氰酸酯封端的2-脲-4[h]-嘧啶酮,记为upy-nco;
[0081]
将所述upy-nco(12.60g,43mmol)、2-氨基-2-甲基-1,3-丙二醇(ampd) (7.03g,66.9mmol)和450ml无水氯仿置于装有冷凝管的圆底烧瓶中,在n2气氛、60℃条件下,回流10h,直至反应完全后,将所得乳白色浑浊溶液真空抽滤,使用大量氯仿洗涤3次,再将所得粉末溶于dmf中,采用离心分离(9000r/min,10min)后取上清液,向上清液中倒入1000ml乙醚进行沉淀,抽滤,真空干燥,得到upy-ampd,即t型扩链剂。
[0082]
实施例1
[0083]
(1)称取10.00g分子量为1000g/mol的ptmg-1000(10mmol),再量取 15mldmac,将两者混合后置于三口烧瓶中,在110℃油浴条件、n2氛围下搅拌1h,得到混合液;
[0084]
(2)待上述混合液温度冷却至80℃后,在n2气氛下,加入ipdi(4.45g, 20mmol)和0.1mmol(0.063g)dbtdl催化剂,继续搅拌4h后,得到预聚物;
[0085]
(3)称取upy-ampd产物(1.99g,5mmol)、量取20ml dmac溶剂,将upy-ampd溶于dmac中,超声溶解后,向预聚物中滴加upy-ampd溶液,80℃搅拌2h,直至扩链反应完成;
[0086]
(4)将所得产物冷却至室温,滴加d230(1.15g,5mmol),在40℃、n2气氛条件下,继续搅拌3h,,将所得产物倒入四氟模具中,并置于真空干燥箱中,在80℃干燥48h,以完全挥发溶剂,得到聚氨酯透明薄膜样品,记为 spu-upy,结构式如式i所示,n=13。
[0087]
实施例2
[0088]
(1)称取ptmg-1000(10.00g,10mmol),再量取15ml dmac,将两者混合后置于三口烧瓶中,在110℃油浴条件、n2氛围下搅拌1h,得到混合液;
[0089]
(2)待上述混合液温度冷却至80℃后,在n2气氛下,加入ipdi(4.45g, 20mmol)和0.1mmol(0.063g)dbtdl催化剂,继续搅拌4h,得到预聚物;
[0090]
(3)称取upy-ampd产物(1.99g,5mmol)、量取20ml dmac溶剂,将upy-ampd溶于dmac中,超声溶解后,向预聚物中滴加upy-ampd溶液,80℃搅拌2h,直至扩链反应完成,得到聚氨酯产物;
[0091]
(4)称取zncl2固体(0.68g,5mmol),再量取5ml dmac溶剂,将zncl2溶于dmac中,超声溶解后,向聚氨酯产物中滴加zncl2溶液,40℃搅拌 5h,进行配位;
[0092]
(5)将配位所得产物冷却至室温,滴加d230(1.15g,5mmol),在40℃、 n2气氛条件
下,再继续搅拌3h,将所得产物倒至四氟模具中,并在真空干燥箱中,在80℃干燥48h,得到超分子聚氨酯弹性体样品,记为spu-upy-zn-1。
[0093]
实施例3
[0094]
(1)称取ptmg-1000(10.00g,10mmol),再量取15ml dmac,将两者混合后置于三口烧瓶中,在110℃油浴条件、n2氛围下搅拌1h,得到混合液;
[0095]
(2)待上述混合液温度冷却至80℃后,在n2气氛下,加入ipdi(4.45g, 20mmol)和0.1mmol(0.063g)dbtdl催化剂,继续搅拌4h,得到预聚物;
[0096]
(3)称取upy-ampd产物(1.99g,5mmol)、量取20ml dmac溶剂,将 upy-ampd溶于dmac中,超声溶解后,向预聚物中滴加upy-ampd混合液,80℃再搅拌2h,直至扩链反应完成,得到聚氨酯产物;
[0097]
(4)称取zncl2固体(0.34g,2.5mmol),再量取5ml dmac溶剂,将zncl2溶于dmac中,超声溶解后,向聚氨酯产物中滴加zncl2溶液,40℃再搅拌 5h,进行配位;
[0098]
(5)将配位所得产物冷却至室温,滴加d230(1.15g,5mmol),在40℃、 n2气氛条件下,再继续搅拌3h,将所得产物倒至四氟模具中,并在真空干燥箱中,在80℃干燥48h,得到超分子聚氨酯弹性体样品,记为spu-upy-zn-2。
[0099]
实施例4
[0100]
(1)称取ptmg-1000(10.00g,10mmol),再量取15ml dmac,将两者混合后置于三口烧瓶中,在110℃油浴条件、n2氛围下搅拌1h,得到混合液;
[0101]
(2)待上述混合液温度冷却至80℃后,在n2气氛下,加入ipdi(4.45g, 20mmol)和0.1mmol(0.063g)dbtdl催化剂,继续搅拌4h后,得到预聚物;
[0102]
(3)称取upy-ampd产物(1.99g,5mmol)、量取20ml dmac溶剂,将 upy-ampd溶于dmac中,超声溶解后,向预聚物中滴加upy-ampd溶液,80℃再搅拌2h,直至扩链反应完成,得到聚氨酯产物;
[0103]
(4)称取zncl2固体(0.17g,1.67mmol),再量取5ml dmac溶剂,将 zncl2溶于dmac中,超声溶解后,向聚氨酯产物中滴加zncl2溶液,40℃再搅拌5h,进行配位
[0104]
(5)将配位所得产物冷却至室温,滴加d230(1.15g,5mmol),在40℃、 n2气氛条件下,再继续搅拌3h,将所得产物倒至四氟模具中,并在真空干燥箱中,在80℃干燥48h,得到超分子聚氨酯弹性体样品,记为spu-upy-zn-3。
[0105]
对比例1
[0106]
(1)称取10.00g分子量为1000g/mol的ptmg-1000(10mmol),再量取 15mldmac,将两者混合后置于三口烧瓶中,在110℃油浴条件、n2氛围下搅拌1h,得到混合液;
[0107]
(2)待上述混合液温度冷却至80℃后,在n2气氛下,加入ipdi(4.45g, 20mmol)和0.1mmol(0.063g)dbtdl催化剂,继续搅拌4h后,将所得产物冷却至室温,滴加d230(10mmol),在40℃、n2气氛条件下,继续搅拌3h,得到聚氨酯样品,记为spu-upy0。
[0108]
对比例2
[0109]
(1)称取10.00g分子量为1000g/mol的ptmg-1000(10mmol),再量取 15mldmac,将两者混合后置于三口烧瓶中,在110℃油浴条件、n2氛围下搅拌1h,得到混合液;
[0110]
(2)待上述混合液温度冷却至80℃后,在n2气氛下,加入ipdi(4.45g, 20mmol)和0.1mmol(0.063g)dbtdl催化剂,继续搅拌4h后,得到预聚物;
[0111]
(3)称取upy-ampd产物(3.98g,10mmol)、量取20ml dmac溶剂,将upy-ampd溶于dmac中,超声溶解后,向预聚物中滴加upy-ampd溶液,80℃搅拌2h,直至扩链反应完成,得到聚氨酯样品,记为spu-upy
1.0
。
[0112]
实施例1~4和对比例1~2的原料配比见表1:
[0113]
表1实施例1~4和对比例1~2原料配比
[0114][0115][0116]
表征及性能测试
[0117]
1)对所制备的upy-nco和upy-ampd进行红外测试,所得结果见图 2;由图2可知,3333cm-1
处的特征峰为t型扩链剂(upy-ampd)中的羟基(-oh-)的伸缩振动峰,2272cm-1
处的特征峰为单体upy-nco的异氰酸酯基团(-nco-)所对应的伸缩振动峰,可初步证明upy-nco和upy-ampd 的成功合成。
[0118]
2)对实施例1制备的聚氨酯以及实施例2~4制备的超分子聚氨酯弹性体进行红外表征,所得结果见图3,其中a为实施例1制备的聚氨酯以及实施例2~4制备的超分子聚氨酯弹性体的红外谱图,b为局部放大图。由图3 可知,图中1708cm-1
归属于自由和无序的氢键-氨基甲酸酯羰基(-c=o-) 的伸缩振动峰,而1642cm-1
处的特征峰为有序氢键-脲羰基(-c=o-)的伸缩振动峰。对比图2中spu-upy和spu-upy-zn的红外光谱可知,随着锌离子的含量的增加,1642cm-1
处的峰强度显著降低,即游离和有序的氢键
‑ꢀ
脲羰基的伸缩振动峰都逐渐消失,并在1676cm-1
处出现了一个新的伸缩振动峰,这表明该体系中的脲羰基(-c=o-)不仅参与形成氢键作用,还与zn
2
配位形成金属配位键。
[0119]
此外,图3中1599cm-1
处的特征峰为脲基嘧啶酮基团中(-cn-)的伸缩振动峰。由于锌离子的掺入,该峰的位置移至1620cm-1
处,故1620cm-1
处的新峰归属于脲基嘧啶酮基团中(-cn-)的移位,也进一步证实了 spu-upy-zn弹性体中脲基嘧啶酮基团中(-cn-)也参与了zn
2
配位形成金属配位键。
[0120]
3)对实施例1制备的spu-upy进行应力-应变曲线测试(测试标准: gb/t 1040-2006,试验速度:10mm/min,试验环境:25℃),并与对比例1~2 制备的spu-upy0和spu-upy1.0进行对比,所得结果见图4;对实施例1 制备的spu-upy以及实施例2~4制备的spu-upy-zn-1、spu-upy-zn-2和 spu-upy-zn-3进行应力-应变曲线测试,所得结果见图5,结果
汇总在表2 中。
[0121]
由图4可知,spu-upy弹性体的拉伸强度随着四重氢键upy含量的增加,应力提高,应变呈下降趋势,即拉伸强度显著增加,断裂应变降低。spu-upy
1.0
的强度最大,可达到11.09mpa。spu-upy弹性体的断裂伸长率呈下降趋势,且spu-upy0的最大值可达4033.26%,而spu-upy
1.0
仅为 131.71%。这可能是由于聚合物网络结构中,氢键作用的增强对聚合物链的力学性能有着显著影响。
[0122]
由图5可知,spu-upy-zn弹性体的拉伸强度随着zn
2
与upy比例的增加而增强,且spu-upy-zn-1的最大值可达14.15mpa。spu-upy-zn弹性体的断裂伸长率随着zn
2
与upy-ampd的比例的增加而降低,spu-upy-zn-3 的最大值为813.53%。即材料应力增加,应变反之减少。因此,zn
2
和脲基嘧啶酮之间引入的配位作用可以显著提高聚氨酯弹性体的拉伸强度和韧性。此外,spu-upy-zn-1弹性体表现出最高的拉伸强度和韧性,分别为14.15 mpa和47.57mj m-3
。结果表明,锌离子的掺入和含有四重氢键的t型扩链剂的含量对聚合物的机械性能有着显著影响。
[0123]
表2不同案例制备的聚氨酯的断裂伸长率、极限抗拉强度和韧性
[0124][0125]
结果表明,由于有效的能量耗散,非共价聚氨酯在外力作用下表现出显著的机械增韧效果。拉伸试验表明,该材料的强度和韧性的增强源于动态和密集的氢键相互作用,这导致了耗散能量的刚性相的连续形成。此外,所引入的zn
2
配位键可以限制流动相的迁移性并增强了链段的结晶度与网络密度,可以有效调节聚氨酯的机械强度。
[0126]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。