1.本发明涉及薯蓣皂苷分子的检测领域,具体涉及一种薯蓣皂苷分子印迹微球、固相萃取装置及制备方法。
背景技术:
2.甾体皂苷类化合物是中药有效成分的重要组成部分,然而由于天然产物的复杂性,植物中甾体皂苷类化合物往往浓度极低,且存在大量的共生组分,其分析过程需要使用繁琐且复杂前处理过程以降低基质干扰。
3.为了降低基质干扰,天然组分固相萃取过程多采用球型树脂填料进行预处理。但商品化的填料对皂苷类缺少特异性识别,在吸附目标物质的同时也会吸附其他结构类似物或性质相近的化合物。
4.为了提高固相萃取填料柱效果,固相萃取填料多采用球型聚合物填料,但球型聚合物在制备过程中需要加入乳化剂以改善形貌,而乳化剂的残留可能会干扰皂苷类化合物的分析结果。
技术实现要素:
5.为了克服上述现有技术中的缺点,本发明的目的在于提供一种薯蓣皂苷分子印迹微球、固相萃取装置及制备方法,所述固相萃取装置采用的薯蓣皂苷分子印迹微球填料对薯蓣皂苷分子的特异性强、不易受干扰。
6.本发明通过以下技术方案实现:
7.一种薯蓣皂苷分子印迹微球的制备方法,包括:
8.s1,将薯蓣皂苷元和甲基丙烯酸溶解在甲苯溶剂中,超声分散,之后加入乙二醇二甲基丙烯酸酯及偶氮二异丁腈,通入氮气除氧,得到预聚合溶液;
9.s2,水中加入二氧化硅微粒及分散助剂,超声分散均匀,热处理,得到二氧化硅溶液;
10.s3,将预聚合溶液和二氧化硅溶液混合,搅拌分散,加热进行聚合反应,聚合反应完成后使用甲醇沉降所得聚合物,采用洗脱液洗除薯蓣皂苷元后,水洗至中性,干燥得到薯蓣皂苷分子印迹微球。
11.优选的,s1中,薯蓣皂苷元、甲基丙烯酸、乙二醇二甲基丙烯酸酯和偶氮二异丁腈比例为41.4mg:(158~264)μl:(1710~2090)μl:20mg。
12.优选的,s2中,分散助剂为triton x-100。
13.优选的,s2中,二氧化硅微粒采用法制备得到。
14.优选的,s3中,聚合反应温度为60~70℃,时间为12~20h。
15.优选的,二氧化硅与薯蓣皂苷元的比例为(30~70)mg:41.4mg。
16.优选的,s3中,搅拌速率为8000~20000rpm。
17.采用所述的制备方法得到的薯蓣皂苷分子印迹微球。
18.一种薯蓣皂苷分子印迹固相萃取装置,包括固相萃取柱,固相萃取柱中的填料为所述的薯蓣皂苷分子印迹微球。
19.与现有技术相比,本发明具有如下的有益效果:
20.本发明方法制备得到的可用于在线固相萃取的薯蓣皂苷分子印迹微球,由于该聚合物制备过程中添加的模板分子薯蓣皂苷与功能单体甲基丙烯酸相互作用,在交联剂乙二醇二甲基丙烯酸酯及引发剂偶氮二异丁腈的作用下模板分子薯蓣皂苷与功能单体之间的相互作用关系被固定下来,在洗脱模板分子薯蓣皂苷之后,便留下与模板分子结构相匹配的孔穴结构,因此该聚合物对薯蓣皂苷具有特异性的识别能力,能够从众多结构化合物中选择性地识别并吸附薯蓣皂苷。进而降低天然产物中其他共存物质干扰。本发明方法制备的分子印迹微球在制备过程中采用二氧化硅微粒作为乳化剂,不需要添加十二烷基磺酸钠等小分子表面活性剂,可以有效避免常规聚合物材料在应用过程中小分子乳化剂残留对薯蓣皂苷分子分析产生的干扰。
21.进一步的,限定薯蓣皂苷元、甲基丙烯酸、乙二醇二甲基丙烯酸酯和偶氮二异丁腈比例为41.4mg:(158~264)μl:(1710~2090)μl:20mg。甲基丙烯酸及乙二醇二甲基丙烯酸酯过低,聚合物吸附量低,过高印迹因子降低。
22.进一步的,采用的分散助剂有利于聚合过程中乳液的稳定。
23.进一步的,聚合反应温度为60~70℃,时间为12~20h,温度过低或时间过短,聚合物无法成形,温度过高或时间过长,聚合物印迹因子降低。
24.进一步的,搅拌速率影响微球的粒径,当搅拌速度低于8000rpm时,所得聚合物粒径分布差异大,形态较差。当转速高于20000rpm时,所得微球粒径过低,不利用后续处理。
25.本发明得到的薯蓣皂苷分子印迹微球对薯蓣皂苷分子特异性强,不易受其他物质干扰。
26.本发明所述在线固相萃取装置构造简单,操作方便、适应性强,有利于提高固相萃取效率并降低天然产物中共存物质干扰,同时避免常规聚合物材料中乳化剂对分析产生的干扰。该装置具有简便快捷、自动化程度高、特异性强、不易受干扰且可重复利用的优点。
附图说明
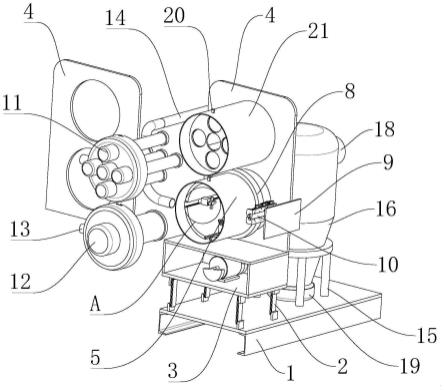
27.图1为薯蓣皂苷分子印迹在线固相萃取装置。
28.图2为实施例1薯蓣皂苷分子印迹微球填料的扫描电子显微镜图,其中图2a为放大100倍,其中图2b为放大1000倍。
29.图3为实施例1薯蓣皂苷分子印迹微球的n2吸附-脱附等温线。
30.图4为实施例1薯蓣皂苷分子印迹微球的吸附动力学曲线。
31.图5为实施例1薯蓣皂苷分子印迹微球的等温吸附曲线。
32.图6为实施例1对不同苷类化合物的识别选择性性结果。
33.图7为实施例1重复使用后吸附量的变化。
34.图8为实施例1~3功能单体优化结果。
35.图9为实施例1~3交联度优化结果。
36.图10为实施例1~4反应温度优化结果。
37.图11为实施例1~4反应时间优化结果。
38.图12为实施例1,4二氧化硅用量优化结果。
39.图13为实施例1,6~7搅拌速度优化结果。
具体实施方式
40.为了进一步理解本发明,下面结合实施例对本发明进行描述,这些描述只是进一步解释本发明的特征和优点,并非用于限制本发明的权利要求。
41.一种薯蓣皂苷分子印迹微球的制备方法,包括:
42.1)制备二氧化硅微粒;
43.2)将薯蓣皂苷元、甲基丙烯酸溶解在甲苯溶剂以形成油相,低温超声分散预组装,之后加入乙二醇二甲基丙烯酸酯及偶氮二异丁腈,溶解后氮气除氧,得到预聚合溶液。
44.3)水中加入二氧化硅微粒及分散助剂triton x-100,超声分散均匀,恒温水浴,得到二氧化硅溶液。
45.4)将步骤2)中得到的预聚合溶液加入到步骤3)中得到的二氧化硅溶液中,剧烈搅拌分散。恒温水浴反应,除去浮于表层的泡沫后使用甲醇沉降聚合物。洗
46.脱液洗除薯蓣皂苷元后,水洗至中性,60℃干燥得到薯蓣皂苷分子印迹微球。
47.所述步骤1)中二氧化硅微粒采用法制备,具体:烧瓶中加入15~20ml水,10~12ml氨水和40~60ml甲醇在磁力搅拌器上进行搅拌。另精密量取50~60ml甲醇和5~7ml teos,进行混合后快速加入上述溶液。25℃下搅拌10~12小时。超高速离心分离。无水乙醇和去离子水洗涤后,50℃下真空干燥。
48.所述步骤2)中薯蓣皂苷元、甲基丙烯酸、乙二醇二甲基丙烯酸酯和偶氮二异丁腈比例为41.4mg:(158~264)μl:(1710~2090)μl:20mg。
49.所述步骤3)中二氧化硅与triton x-100的加入比例为(30~70)mg:0~20μl。二氧化硅与薯蓣皂苷元的比例为(30~70)mg:41.4mg。
50.所述步骤4)中搅拌速率为8000~20000rpm,恒温水浴温度为60~70℃,反应时间为12~20h。
51.薯蓣皂苷分子印迹在线固相萃取装置的构建:
52.将所得薯蓣皂苷分子印迹微球分散于甲醇中并装填于在线固相萃取柱中,在线固相萃取柱的装填量为100~400mg,使用甲醇洗涤,微量注射泵通过三通阀与固相萃取柱相连,固相萃取柱通过十通切换阀与液相色谱相连,构建得到固相萃取在线分析装置。
53.实施例1:一种薯蓣皂苷分子印迹微球制备方法
54.1)在250ml烧瓶中加入17ml水、11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空干燥24小时。
55.2)41.4mg薯蓣皂苷元、176μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入1900μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液;
56.3)10ml的水中加入20μl的triton x-100,随后加入60mg的sio2,超声分散,以形成稳定的悬浮液,得到sio2溶液维持温度在70℃。
57.4)在sio2溶液中加入预聚合溶液,使用16000rpm均质乳化以获得稳定的pickering乳液。60℃聚合16小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷元被检测到。水洗至中性后60℃干燥得薯蓣皂苷分子印迹聚合物(mips)。
58.空白分子印迹聚合物(nips)的合成与洗脱方法与此相同,只是在合成过程中未加入模板分子薯蓣皂苷元。
59.将200mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
60.本实施例的有益效果为:
61.参照图2,从图2可以看出,用实施例1制备得到的薯蓣皂苷分子印迹微球粒径相对均匀,其平均粒径在44.31μm左右。同时微球表面较为粗糙,为识别提供了吸附位点。
62.参照图3,从图3可以看出,用实施例1制备得到的薯蓣皂苷分子印迹聚合物呈现出ⅳ型等温线,意味薯蓣皂苷分子印迹聚合物属于大孔材料。薯蓣皂苷分子印迹聚合物滞后环上下两端闭合点相距更远,表明薯蓣皂苷分子印迹聚合物表面的孔径分布范围比空白分子印迹聚合物更宽,更加粗糙。
63.参照图4,从图4可以看出,用实施例1制备得到的薯蓣皂苷分子印迹聚合物传质速率快,在大约10分钟以内就可以实现吸附平衡,平衡吸附量为42.6mg/g。
64.参照图5,从图5可以看出,用实施例1制备得到的随着母液浓度的增加,薯蓣皂苷分子印迹聚合物的饱和吸附量不断增大,当母液浓度为200μg/ml时,薯蓣皂苷分子印迹聚合物的饱和吸附量最大,达到43.1mg/g。
65.参照图6,从图6可以看出,用实施例1制备得到的薯蓣皂苷分子印迹聚合物对薯蓣皂苷的吸附量最大,吸附量为43mg/g,印迹因子为7.86。
66.参照图7,从图7可以看出,用实施例1制备得到的薯蓣皂苷分子印迹聚合物在重复使用5次后,其对薯蓣皂苷的吸附量依旧维持在40mg/g以上,表明该薯蓣皂苷分子印迹聚合物耐用性良好。
67.实施例2:一种薯蓣皂苷分子印迹微球的构建方法
68.1)在250ml烧瓶中加入17ml水,11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空干燥24小时。
69.2)41.4mg薯蓣皂苷元、158μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入1710μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液。
70.3)10ml的水中加入60mg的sio2,超声分散,以形成稳定的悬浮液,得到二氧化硅溶
液,维持温度在70℃。
71.4)在二氧化硅溶液中加入预聚合溶液,使用16000rpm均质乳化以获得稳定的pickering乳液。70℃聚合20小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷元被检测到。水洗至中性后60℃干燥。
72.将100mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取空柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
73.实施例3:一种薯蓣皂苷分子印迹微球制备方法
74.1)在250ml烧瓶中加入17ml水、11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空干燥24小时。
75.2)41.4mg薯蓣皂苷元、264μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入2090μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液;
76.3)10ml的水中加入20μl的triton x-100,随后加入60mg的sio2,超声分散,以形成稳定的悬浮液,得到sio2溶液维持温度在70℃。
77.4)在sio2溶液中加入预聚合溶液,使用16000rpm均质乳化以获得稳定的pickering乳液。60℃聚合16小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷元被检测到。水洗至中性后60℃干燥。
78.将400mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
79.参照图8,从实施例1~3合成过程中功能单体甲基丙烯酸含量对薯蓣皂苷的吸附的影响的柱状图可以看出,随着甲基丙烯酸加入量的增加,薯蓣皂苷分子印迹聚合物的吸附量逐渐增大,在甲基丙烯酸用量为176μl时吸附量及印迹因子最大,继续增加甲基丙烯酸用量,吸附量变化不大,但印迹因子降低。
80.参照图9,从实施例1~3合成过程中交联剂乙二醇二甲基丙烯酸酯含量对薯蓣皂苷的吸附的影响的柱状图可以看出,随着乙二醇二甲基丙烯酸酯加入量的增加,薯蓣皂苷
分子印迹聚合物的吸附量逐渐增大,在乙二醇二甲基丙烯酸酯用量为1900μl时吸附量及印迹因子最大,继续增加乙二醇二甲基丙烯酸酯用量,吸附量变化不大,但印迹因子降低。
81.实施例4:一种薯蓣皂苷分子印迹微球制备方法
82.1)在250ml烧瓶中加入17ml水、11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空干燥24小时。
83.2)41.4mg薯蓣皂苷元、176μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入1900μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液;
84.3)10ml的水中加入20μl的triton x-100,随后加入60mg的sio2,超声分散,以形成稳定的悬浮液,得到sio2溶液维持温度在70℃。
85.4)在sio2溶液中加入预聚合溶液,使用16000rpm均质乳化以获得稳定的pickering乳液。60℃聚合12小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷被检测到。水洗至中性后60℃干燥。
86.将200mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
87.参照图10,从实施例1~4合成过程中反应温度对薯蓣皂苷的吸附的影响的柱状图可以看出,随着反应温度的增加,薯蓣皂苷分子印迹聚合物的吸附量逐渐增大,但印迹因子降低。在60℃时达到最佳。
88.参照图11,从实施例1~4合成过程中反应温度对薯蓣皂苷的吸附的影响的柱状图可以看出,随着反应时间的增加,薯蓣皂苷分子印迹聚合物的吸附量轻微增大,印迹因子轻微降低。在16h时达到最佳。
89.实施例5:一种薯蓣皂苷分子印迹微球制备方法
90.1)在250ml烧瓶中加入17ml水、11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空干燥24小时。
91.2)41.4mg薯蓣皂苷元、176μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入1900μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液;
92.3)10ml的水中加入20μl的triton x-100,随后加入30mg的sio2,超声分散,以形成稳定的悬浮液,得到sio2溶液维持温度在70℃。
93.4)在sio2溶液中加入预聚合溶液,使用16000rpm均质乳化以获得稳定的pickering乳液。60℃聚合16小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷元被检测到。水洗至中性后60℃干燥。
94.将200mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
95.参照图12,从实施例1、4合成过程中二氧化硅的加入量影响着微球的粒径,随着二氧化硅加入量的增加,微球粒径降低,均匀性增强,当二氧化硅加入量大于60mg时,粒径及均匀度基本维持恒定不变。
96.实施例6:一种薯蓣皂苷分子印迹微球制备方法
97.1)在250ml烧瓶中加入17ml水、11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空干燥24小时。
98.2)41.4mg薯蓣皂苷元、176μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入1900μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液;
99.3)10ml的水中加入20μl的triton x-100,随后加入70mg的sio2,超声分散,以形成稳定的悬浮液,得到sio2溶液维持温度在70℃。
100.4)在sio2溶液中加入预聚合溶液,使用20000rpm均质乳化以获得稳定的pickering乳液。60℃聚合16小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷元被检测到。水洗至中性后60℃干燥。
101.将200mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
102.实施例7:一种薯蓣皂苷分子印迹微球制备方法
103.1)在250ml烧瓶中加入17ml水、11.5ml氨水和50ml甲醇在磁力搅拌器上进行搅拌。另精密量取58ml甲醇和7ml teos,进行混合后快速加进上述溶液,25℃下搅拌10小时。10000r/min离心分离10min。无水乙醇和去离子水洗3次后,将所得sio2颗粒在50℃下真空
干燥24小时。
104.2)41.4mg薯蓣皂苷元、176μl甲基丙烯酸溶解在1800μl甲苯溶剂中,超声仪进行处理5分钟,25℃自组装12h。加入1900μl乙二醇二甲基丙烯酸酯和20mg偶氮二异丁腈,通入氮气15分钟,得到预聚合溶液;
105.3)10ml的水中加入20μl的triton x-100,随后加入60mg的sio2,超声分散,以形成稳定的悬浮液,得到sio2溶液维持温度在70℃。
106.4)在sio2溶液中加入预聚合溶液,使用8000rpm均质乳化以获得稳定的pickering乳液。60℃聚合16小时后,获得了薯蓣皂苷分子印迹微球。除去混合液中的泡沫,甲醇沉降获得微球。将微球用水和乙醇冲洗后,使用甲醇/乙酸(90:10)索氏提取器洗脱至无薯蓣皂苷元被检测到。水洗至中性后60℃干燥。
107.将200mg薯蓣皂苷分子印迹微球与甲醇混合后转移至固相萃取柱中,连续注入甲醇使填料紧实。参照图1构建薯蓣皂苷分子印迹在线固相萃取装置。装置具体包括薯蓣皂苷固相萃取单元(具体可采用固相萃取柱1),阀切单元(具体可采用固相萃取柱2),定量转移单元(具体可采用定量环3),样品/溶剂提供单元(用于提供所需样品或溶剂,具体可采用注射泵4),还包括色谱输液单元(具体可采用色谱泵5),色谱流动相盛放单元(具体可采用溶剂瓶6),色谱分析柱单元(具体可采用c18色谱柱7),检测单元(具体可采用紫外检测器8)及废液收集单元(具体可采用废液瓶9)。
108.参照图13,从实施例1、6~7合成过程搅拌转速影响着微球的粒径,随着转速的增加,微球粒径降低,均匀性增强,当转速达到16000转时,粒径及均匀度基本维持恒定不变。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。