一种化学酶法合成
α-肌营养不良蛋白聚糖相关糖肽的方法
技术领域
1.本发明属于医药技术领域。更具体地,本发明利用化学酶法高效地合成了α-肌营养不良蛋白聚糖相关糖肽。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.肌营养不良蛋白聚糖(dystroglycan,dg)是一种跨膜糖蛋白,dg蛋白是抗肌萎缩蛋白糖蛋白复合体(dystrophin-glycoprotein complex,dgc)的一个重要的部分,dg由两个单个基因编码的α-和β-亚基组成,分别命为α-肌营养不良蛋白聚糖(α-dg)和β-肌营养不良蛋白聚糖(β-dg)。dg基因在骨骼肌等多的细胞的表达中起着至关重要的作用。dg基因突变本身未发现与疾病有关,但是翻译后修加工(主要是糖基化)水平的改变会导致肿瘤等重大疾病。
4.报道称,β-肌营养不良蛋白聚糖(β-dg)能参与骨骼肌中多种信号转导,但作用机制尚不明确。α-肌营养不良蛋白聚糖(α-dg)主要存在于骨骼肌、神经和大脑的细胞膜中,它在连接细胞骨架和细胞外基质的过程中起着至关重要的作用,在维护肌肉完整性等各种生命活动中必不可少。因此近年来α-dg受到大家的广泛关注。
5.α-dg表面的正常o-甘露糖基化修饰对其功能具有重要影响。若o-甘露糖基化生物合成途径异常、糖链表达过低,会导致dgc结构的破坏,影响肌肉组织的功能。此外,o-甘露聚糖跟多种病理过程密切相关,如o-甘露糖基化翻译后修饰的缺陷会影响乳腺癌等多种类型癌症的转移;异常的o-甘露糖基化修饰会破坏肌营养不良受体的功能,影响神经系统的生长、发育、修复和信号传递,导致各类先天肌营养不良症(cmds)等。
6.但o-甘露聚糖的结构相对复杂多样,难以从自然界中提取分离到结构明确、足够量、高纯度的o-甘露聚糖,从而影响对α-dg结构和功能的系统理解。目前,o-甘露聚糖的合成方法,主要有化学合成和酶法合成。(1)化学合成法需要进行繁琐的保护基操作,路线较长,糖苷化反应选择性较差,产率较低。(2)酶法合成策略采用的哺乳动物酶的催化效率低、底物适应性差。
7.在糖肽的合成中,也存在诸多挑战。常用的汇聚式合成策略主要针对n-糖链多肽的制备。而本发明中制备的是o-糖链多肽,需要考虑糖链与多肽连接时立体构型的问题,若采用直接连接的方法进行化学反应难度较大;且由于糖链空间位阻较大,导致连接效率较低的问题。
8.因此,大量制备结构确定的高纯度o-甘露聚糖和α-dg相关o-糖链多肽成为目前亟待解决的问题。
技术实现要素:
9.为了解决上述问题,本发明提供了一种化学法合并酶法高效制备o-甘露聚糖和α-dg相关的复杂o-甘露糖肽的方法。
10.为实现上述技术目的,本发明采用如下技术方案:
11.本发明的第一个方面,提供了一种化学酶法合成α-肌营养不良蛋白聚糖相关糖肽的方法,包括:
12.以o-糖基三氯乙酰亚胺酯为糖基供体,通过催化糖苷化方法,合成立体构型专一的o-糖氨基酸;
13.以所述o-糖氨基酸为原料,采用固相多肽合成方法组装糖肽;
14.采用酶法对所述糖肽进行糖链修饰,即得。
15.本发明的第二个方面,提供了上述的方法合成的糖肽,实现结构复杂的糖肽的多样化合成。
16.(1)运用糖基三氯乙酰亚胺酯为糖基供体,通过催化糖苷化方法,合成立体构型专一的o-糖氨基酸,(2)通过微波辅助的固相多肽合成方法,合成带有特定糖链修饰的α-dg相关糖肽,(3)通过酶法进行糖链延伸和修饰,获得结构复杂多样化的α-dg相关糖肽化合物库。
17.本发明的有益效果在于:
18.(1)本发明提供了一种高效的o-甘露聚糖糖氨基酸的制备方法,反应产率高、选择性好。
19.(2)本发明提供了一种高效的o-糖肽的制备方法,适用于结构复杂的o-甘露糖肽的多样化制备。
20.(3)本技术的操作方法简单、实用性强、具有普适性,易于规模化生产。
附图说明
21.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
22.图1.化合物4核磁谱图;
23.图2.化合物6核磁谱图;
24.图3.化合物7核磁谱图;
25.图4.糖肽16:hplc图和质谱图
26.图5.糖肽17:hplc图和质谱图
27.图6.糖肽18:hplc图和质谱图
28.图7.糖肽19:hplc图和质谱图
29.图8.糖肽20:hplc图和质谱图
30.图9.糖肽21:hplc图和质谱图
31.图10.糖肽22:hplc图和质谱图
32.图11.糖肽23:hplc图和质谱图
33.图12.糖肽24:hplc图和质谱图
34.图13.糖肽25:hplc图和质谱图
35.图14.糖肽26:hplc图和质谱图
36.图15.糖肽27:hplc图和质谱图
具体实施方式
37.应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
38.一种化学酶法合成α-肌营养不良蛋白聚糖相关糖肽的方法,其特征在于,将化学方法与酶法相结合,具体包括以下步骤:以化学方法合成立体构型专一的糖链结构的糖氨基酸,通过固相多肽合成方法组装糖肽,通过酶法进行糖链修饰,实现结构复杂的糖肽的多样化合成。
39.在一些实施例中,所述o-糖氨基酸为o-甘露糖,core m1型o-甘露聚糖,core m2型o-甘露聚糖,core m3型o-甘露聚糖。
40.在一些实施例中,氨基酸结构为丝氨酸或苏氨酸。
41.在一些实施例中,所述催化糖苷化采用的催化剂为酸性催化剂。
42.在一些实施例中,所述催化剂为tmsotf、bf3·
oet2或tfoh。
43.在一些实施例中,所述固相多肽合成在微波辅助、氮气鼓泡下进行。
44.在一些实施例中,所述的糖链修饰的方法为半乳糖基化或唾液酸化。
45.在一些实施例中,糖基化位点可为一个或多个。
46.下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
47.实施例1
48.1.化学合成o-甘露糖基化糖氨基酸
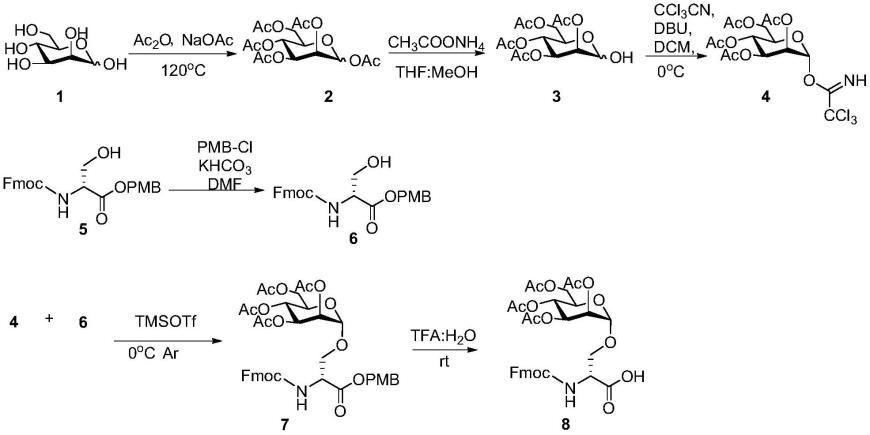
49.1.1甘露糖-丝氨酸(man-ser)糖氨基酸的合成路线
[0050][0051]
具体的实验步骤如下:
[0052]
1,2,3,4,6-五-氧-乙酰基-d-吡喃甘露糖(2)的制备
[0053]
在氮气的保护下,在单口圆底烧瓶中加入适量的醋酸酐溶解甘露糖1(30g,
0.16mol,1.0eq),加入乙酸钠(34g,0.42mol,2.5eq),加热温度至120℃,回流2小时,磁力搅拌,反应完全后,用饱和碳酸氢钠水溶液萃取,无水硫酸钠干燥,旋干,几乎定量得到粗品五乙酰基甘露糖2。
[0054]
2,3,4,6-四-o-乙酰基-1-氧-β-d-吡喃甘露糖(3)的制备
[0055]
将化合物2(35g,89.7mmol,1.0eq)和乙酸铵(ch3coonh4)(34.6g,449mmol,5eq)溶于200ml thf和meoh(thf:meoh=1:1,v/v)的混合溶液中,室温搅拌12小时,tlc检测原料反应完全后,旋蒸干溶剂,用饱和氯化钠水溶液和二氯甲烷萃取,柱层析分离纯化(石油醚:乙酸乙酯=1:1)得白色固体3(21.4g,产率69%)。
[0056]
2,3,4,6-四-氧-乙酰基-1-氧-β-d-吡喃甘露糖基三氯乙酰亚胺酯(4)的制备
[0057]
将化合物3(6.7g,192.5mmol,1.0eq)溶于50ml干燥的二氯甲烷中,在0℃、氮气保护的条件下,向该反应体系中依次加入三氯乙腈(5.8ml,57.7mmol,3eq)和1,8-二氮杂二环十一碳-7-烯(dbu)(0.3ml,1.93mmol,0.1eq),让其自然升至室温,搅拌2小时,tlc检测原料反应完全,旋蒸干溶剂后,经柱层析分离(石油醚:乙酸乙酯=1:1)得白色粘稠油状物4(8.3g,产率89%)。
[0058]nα-(9-芴甲氧羰基)-o-对甲氧基苄基-l-丝氨酸(6)的制备
[0059]
将化合物5(1g,3.1mmol,1.0eq)和碳酸氢钾(khco3)(0.6g,6.1mmol,2.0eq)溶于20ml干燥的n,n-二甲基甲酰胺(dmf)中,室温下搅拌,加入四丁基碘化铵(tbai)(0.11g,0.31mmol,0.1eq),缓慢滴加4-甲氧基苄氯(pmbcl)(828μl,6.1mmol,2.0eq),室温搅拌12小时,tlc检测原料反应完全,蒸干溶剂,残留物用50ml二氯甲烷溶解,依次用50ml1mol/l盐酸溶液,50ml饱和碳酸氢钠溶液,50ml饱和氯化钠水溶液洗涤,有机相用无水硫酸钠干燥,抽滤除去干燥剂,滤液浓缩后经柱层析分离(石油醚:乙酸乙酯=1:1,v/v)得白色固体6(1.1g,产率82%)。
[0060]nα-(9-芴甲氧羰基)-2,3,4,6-四-o-乙酰基-α-d-吡喃甘露糖基-l-丝氨酸对甲氧基苄基酯(7)的制备
[0061]
在氩气保护的条件下,称取化合物4(640mg,1.30mmol,1.0eq)和化合物6(644mg,1.43mmol,1.1eq)于装有已经活化好的分子筛的两口瓶中,加入干燥的二氯甲烷5ml溶解,在0℃下,加入三氟甲磺酸三甲基硅脂(tmsotf)(47.2μl,0.261mmol,0.2eq),反应10min后,tlc检测原料反应完全,用三乙胺淬灭反应。滤去分子筛,蒸干溶剂,柱层析分离纯化(二氯甲烷:乙酸乙酯=6:1)得到糖苷化产物7(874mg,产率86%)。
[0062]nα-(9-芴甲氧羰基)-2,3,4,6-四-o-乙酰基-α-d-吡喃甘露糖基-l-丝氨酸(8)
[0063]
将化合物7(350mg,0.45mmol,1eq)溶于三氟乙酸(2.999ml,40.5mmol,90eq)与水(tfa:h2o=19:1)的混合溶液中,室温下搅拌10min,tlc(乙酸乙酯:甲醇=5:1,v/v)检测显示反应完全,dcm稀释,饱和碳酸氢钠溶液萃取,无水硫酸钠干燥,旋干得白色固体8,直接用于固相合成。
[0064]
1.2.o-甘露聚糖core m1、core m2和core m3三个核心结构糖氨基酸底物的制备
[0065]nα-(9-芴甲氧羰基)-o-2-乙酰氨基-3,4,6-三-o-乙酰基-2-脱氧-β-d-吡喃葡萄糖基-(1
→
2)-3,4,6-三-o-乙酰基-α-d-吡喃甘露糖基-l-丝氨酸(10)
[0066][0067]
将化合物9(480mg,0.45mmol,1eq)溶于三氟乙酸(3.076ml,40.56mmol,90eq)与水(tfa:h2o=19:1)的混合溶液中,室温下搅拌10min,tlc(乙酸乙酯:甲醇=5:1,v/v)检测显示反应完全,dcm稀释,饱和碳酸氢钠溶液萃取,无水硫酸钠干燥,旋干得白色固体10,直接用于固相合成。
[0068]nα-(9-芴甲氧羰基)-o-[2-乙酰氨基-3,4,6-三-o-乙酰基-2-脱氧-β-d-吡喃葡萄糖基-(1
→
2)-3,4,6-三-o-乙酰基-2-乙酰氨基-β-d-吡喃葡萄糖基-(1
→
6)-3,4-二-o-乙酰基-1-o-α-d-吡喃甘露糖基]-l-丝氨酸(12)
[0069][0070]
将化合物11(68mg,0.05mmol,1eq)溶于三氟乙酸(0.335ml,4.5mmol,90eq)与水(tfa:h2o=19:1)的混合溶液中,室温下搅拌10min,tlc(乙酸乙酯:甲醇=5:1,v/v)检测显示反应完全,dcm稀释,饱和碳酸氢钠溶液萃取,无水硫酸钠干燥,旋干得白色固体12,直接用于固相合成。
[0071]nα-(9-芴甲氧羰基)-o-[2-乙酰氨基-3,4,6-三-o-乙酰基-2-脱氧-β-d-吡喃葡萄糖基-(1
→
4)-2-o-苯甲酰基-3,6-二-o-乙酰基-1-o-α-d-吡喃甘露糖基]-l-丝氨酸(14)
[0072][0073]
将化合物13(26.6mg,0.05mmol,1eq)溶于三氟乙酸(333.2ml,4.5mmol,90eq)与水(tfa:h2o=19:1)的混合溶液中,室温下搅拌10min,tlc(乙酸乙酯:甲醇=5:1,v/v)检测显示反应完全,dcm稀释,饱和碳酸氢钠溶液萃取,无水硫酸钠干燥,旋干得白色固体14,直接
用于固相合成。
[0074]
2.α-dg糖肽的固相合成
[0075]
2.1本发明通过化学方法固相合成7条α-dg糖肽(对应命名为15-21),序列见表1
[0076]
表1含有糖氨基酸的多肽序列
[0077][0078]
化学固相合成糖肽的通用方法:
[0079]
(1)称取2-chlorotrityl chloride树脂(树脂负载量为0.5mmol/g)100mg于10ml固相反应管中,加2ml干燥的dcm进行溶胀5min,用干燥的dcm洗三次,每次1ml。
[0080]
(2)称取1.0eq的糖氨基酸(化合物8,10,12和14),用1ml干燥的dcm溶解,加2.5eq的diea,混合均匀。转移至固相反应管中,现象为有白色烟雾产生,室温密封振摇12h,加入甲醇(0.8ml/g resin)密封振摇5min淬灭反应。分别用dcm、dmf溶剂各洗三次,并进行原料糖氨基酸的回收利用。
[0081]
(3)然后将树脂转移至微波反应器的固相反应管中,抽取50%吗啡啉(吗啡啉:dmf=1:1,v/v)2ml,在60℃,25w的条件下,氮气鼓气反应10min,抽干,用dmf溶剂洗三次。
[0082]
(4)称取4.0eq的fmoc-aa-oh(普通氨基酸或糖氨基酸)、3.9eq的hatu于4ml ep管中,加入2ml dmf溶解,再加6eq的diea混合均匀。转移至微波反应器的固相反应管中,在50℃,20w的条件下,氮气鼓气反应10min后,停止反应,用dmf溶剂洗三次。必要时,以相同的条件再重复进行一次偶联。随后,抽取50%吗啡啉(吗啡啉:dmf=1:1,v/v)2ml,在60℃,25w的条件下,氮气鼓气反应10min,抽干,用dmf溶剂洗三次。
[0083]
(5)循环重复上述(4)的操作,直至多肽序列组装完成。
[0084]
(6)量取ac2o,pyr(ac2o:pyr:dmf=0.1:0.1:0.8,v/v/v)共2ml反应20min,停止反应,用dmf、dcm溶剂各洗三次。
[0085]
(7)量取水合肼(dmf:水合肼=0.6:0.4)共2ml反应4小时,将糖上保护基团进行脱除,用dmf、dcm溶剂各洗三次(因dmf不易挥发,所以最后用dcm洗涤)。
[0086]
(8)加入tfa(tfa:tips:h2o=95:2.5:2.5,v/v/v)进行树脂的切割,室温振荡反应约2h。把肽链从树脂上切除下来,将切割液用氮气吹干,加入约25ml冰乙醚沉淀三次,待多肽固体析出后,离心3次,转速4000rpm每次5min,收集沉淀,加入少量mecn/h2o/0.1%ch3cooh溶解后,利用半制备液相对多肽粗品进行分离纯化,收集产物将样品冻干,得到目标糖肽。
[0087]
具体实验步骤如下:
[0088]
化合物15的合成
[0089][0090]
化合物15的合成是以化合物8为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体15(33.7mg,产率:54%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,梯度洗脱5-95%b in a over 15min,λ=220nm,保留时间为6.848min,esi-ms:m/z:[m h]
calcd for c
51h82
n9o
272
1252.53,found:1251.68;[m na]
calcd for c
51h81
n9nao
27
1274.51,found:1274.06。
[0091]
化合物16的合成
[0092][0093]
化合物16的合成是以化合物10为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体16(46.9mg,产率:73%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.930min,esi-ms:m/z:[m h]
calcd for c
53h85n10o27
1293.56,found:1293.32;[m na]
calcd for c
53h84n10o27
1315.54,found:1315.20;[m 2h]
2
calcd for c
53h86n10o272
647.28,found:647.00;[m nh4]
2
calcd for c
53h88n11o272
655.29,found:655.49。
[0094]
化合物17的合成
[0095][0096]
化合物17的合成是以化合物10为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体17(42.1mg,产率:51%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.607min,esi-ms:m/z:[m 2h]
2
calcd for c
67h109n11o372
829.85,found:829.74。
[0097]
化合物18的合成
[0098][0099]
化合物18的合成是以化合物12为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体18(14mg,产率:19%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.748min,esi-ms:m/z:[m 2h]
2
calcd for c
61h99n11o322
748.82,found:748.62;[m nh4]
2
calcd for c
61h101n11o322
756.83,found:756.67。
[0100]
化合物19的合成
[0101][0102]
化合物19的合成是以化合物14为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体19(27.9mg,产率:43%)。通过hplc和esi-ms鉴定,hplc色谱条件:
solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为7.001min,esi-ms:m/z:[m h]
calcd for c
53h85n10o27
1293.56,found:1292.82。
[0103]
化合物20的合成
[0104][0105]
化合物20的合成是以化合物10为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体20(28.5mg,产率:43%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.552min,esi-ms:m/z:[m h]
calcd for c
54h88n11o28
1338.58,found:1337.82;[m 2h]
2
calcd for c
54h89n11o282
669.79,found:669.46。
[0106]
化合物21的合成
[0107][0108]
化合物21的合成是以化合物12为底物,经过化学固相合成糖肽的通用方法,过半制备型液相得白色固体21(28.6mg,产率:37%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.363min,esi-ms:m/z:[m h]
calcd for c
62h101n12o33
1541.66,found:1540.69;[m 2h]
2
calcd for c
62h102n12o332
771.33,found:770.94;[m 2h]
3
calcd for c
62h102n12o332
1028.44,found:1027.25。
[0109]
3.酶法糖链修饰,合成复杂糖肽
[0110]
本发明所用的酶均在大肠杆菌系统中重组表达,并且都含组氨酸标签,所以其表达纯化过程基本相同,将操作步骤如下。
[0111]
3.1养菌提酶
[0112]
3.1.1大肠杆菌的培养
[0113]
(1)培养基的配置及灭菌
[0114]
制备1l培养基和25ml培养基各5个,其中培养基成分为:酵母提取物5g/l、胰蛋白胨10g/l、氯化钠10g/l,按上述各成分含量进行称取并加入到5l烧杯中,加入5.125l双蒸
水,搅拌使其溶解,然后将溶液以每个1l和每个25ml的体积分别分装于2l锥形瓶和100ml锥形瓶中,并将锥形瓶密封好。然后将密封好的摇瓶和各种规格的移液枪枪头置于高压灭菌锅中,121℃条件下灭菌20min,结束后,将报纸包装的枪头盒放于烘箱内烘干,然后转至无菌操作台备用。
[0115]
(2)大肠杆菌的小量复苏
[0116]
在无菌操作台内,首先在100ml摇瓶中加入100mg/ml的氨苄青霉素钠溶液25μl,使其终浓度为100μg/ml,摇匀后,加入菌种保存液50μl,密封好以后放置于摇床中,设置条件37℃,转速225r/min,培养约14h。
[0117]
(3)大肠杆菌的大量培养
[0118]
在无菌操作台内,向1l的摇瓶中加入100mg/ml的氧节青霉素钠溶液1ml,使其终浓度为100μg/ml,摇匀后,加入小量复苏的菌液10ml。在37℃、225r/min条件下培养3-4h,检测600nm波长处的吸光度值(od 600),当od 600在0.6-0.8之间时,进行诱导。
[0119]
(4)酶的诱导表达
[0120]
向大量培养好的菌液中加入1mol/l的iptg溶液100μl,使其终浓度为0.1mmol/l进行诱导,密封好后,置于摇床,设置温度为20℃、转速225r/min,培养18-20h。
[0121]
(5)菌体收集和保存
[0122]
菌液浓度达到理想值后,将菌液置于离心杯中,8000r/min、4℃条件下离心5min,弃去上清液,将底部沉淀菌体收集,保存在-80℃冰箱备用。
[0123]
3.1.2酶的纯化
[0124]
(1)超声破碎
[0125]
将收集到的菌液分多次加入15-20ml裂解液(20mmol/l tris-hcl,ph 8.0,0.1%tritonx-100)进行溶解,并将菌液转移至100ml烧杯中,用超声破碎仪对菌液进行超声,即得裂解菌体。超声使用方法:为保持低温环境,将100ml烧杯固定在冰上进行超声,超声过程要时刻观察,确保超声探头距离烧杯底部1cm左右,太高会影响裂解效果,条件为on 3s,off 5s,超声30min。
[0126]
(2)低温离心
[0127]
将上述的裂解液转移到50ml离心管中配平,在4℃、12000r/rnin条件下离心25min,取上清,进行进一步的纯化。
[0128]
(3)蛋白纯化
[0129]
因为本文所涉及的酶都含有his-tag的,所以纯化过程应用镍离子亲和层析柱(ni
2 -nta)进行酶的纯化,具体步骤如下:
[0130]
a)首先要用10倍柱体积三蒸水冲洗柱子,将柱子中的乙醇保存液冲洗干净。然后用10倍柱体积的结合缓冲液预平衡柱子(binding buffer:5mmol/l imidazole,0.5mol/l nacl,50mmol/l tris-hcl,ph 8.0)。
[0131]
b)上样前,需要将离心后的上清液用0.45μm的水系滤膜过滤,减少杂质对镍柱的损伤。
[0132]
c)上样,并用8个柱体积的结合缓冲液平衡柱子。
[0133]
d)用8个柱体积的清洗缓冲液冲洗柱子(washing buffer:20mmol/l imidazole,0.5mol/l nacl,50mmol/l tris-hcl,ph 8.0),洗脱掉非特异性结合的杂蛋白,直至流出液
加入考马斯亮蓝不显蓝色。
[0134]
e)用8个柱体积的洗脱缓冲液冲洗柱子(eluting buffer:200mmol/l imidazole,0.5mol/l nacl,50mmol/l tris-hcl,ph 8.0),将目的蛋白洗脱下来,分管收集,并用考马斯亮蓝检测。含目的蛋白量较高的洗脱液收集到一起,保存在4℃冰箱,或者加入10%甘油保存在-20℃冰箱。
[0135]
f)最后将镍柱用10个柱体积三蒸水清洗,并保存在20%乙醇中。
[0136]
所有的酶在使用之前都需要进行小试来确定其酶活。
[0137]
3.1.3udp-gal的制备
[0138]
将半乳糖(gal,1.0eq)、三磷酸腺苷(atp,1.5eq)和三磷酸尿苷(utp,1.5eq)溶解在50ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至7.5,最后加入适量的酶,ecgalk和blusp将反应液定容至设定的体积,然后在恒温摇床中以225r/min转速,37℃反应。通过薄层色谱监测反应进程,并用茴香醛染色剂染色。当反应完全后,加入等体积的冰乙醇终止反应,在-20℃冰箱放置30min,离心除去蛋白沉淀,含产物的上清液浓缩后依次过bio-gel p2(纯水洗脱),deae(水、5%1mol/l nacl、15%1mol/l nacl和1mol/l nacl梯度洗脱)和bio-gel p2(纯水洗脱)柱进行纯化。
[0139]
3.1.4cmp-neu5ac的制备
[0140]
将唾液酸(neu5ac,1.0eq)、三磷酸胞苷(ctp,1.5eq)溶解在50ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至8.0,最后加入适量的酶,nmcss。将反应液定容至设定的体积,然后在恒温摇床中以225r/min转速,37℃反应。通过薄层色谱监测反应进程,并用茴香醛染色剂染色。当反应完全后,加入等体积的冰乙醇终止反应,在-20℃冰箱放置30min,离心除去蛋白沉淀,含产物的上清液浓缩后依次过bio-gel p2(纯水洗脱)、deae(水、5%1mol/l nacl、15%1mol/l nacl和1mol/l nacl梯度洗脱)和bio-gel p2(纯水洗脱)柱进行纯化。
[0141]
3.2酶法糖链修饰
[0142]
化合物22的合成
[0143][0144]
将16(10mg)、udp-gal(118.3mg)溶解在4ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至7.5,最后加入酶nmlgtb。加入双蒸水至终体积为5ml。将该反应体系至于恒温摇床中以225r/min转速,37℃反应12h。过半制备型液相得白色固体22(6.5mg,产率:58%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.869min,esi-ms:m/z:[m 2h]
2
calcd for c
59h96n10o322
728.31,found:
728.16;[m nh4]
2
calcd for c
59h98n11o322
736.32,found:736.20。
[0145]
化合物23的合成
[0146][0147]
将22(6mg)、cmp-neu5ac(32.8mg)溶解在4ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至8.5,最后加入酶pmst1 m144d。加入双蒸水至终体积为3ml。将该反应体系至于恒温摇床中以225r/min转速,37℃反应5h。过半制备型液相得白色固体23(3.6mg,产率:49%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent c:25mm nh4oac;solvent d:ch3cn containing 10%25mm nh4oac,5-95%d in c over 15min,λ=220nm,保留时间为6.803min,esi-ms:m/z:[m 2h]
2
calcd for c
70h113n11o402
873.86,found:873.40;[m nh4]
2
calcd for c
70h115n12o402
881.87,found:881.58,[m 2nh4]
2
calcd for c
70h119n13o402
890.88,found:890.18。
[0148]
化合物24的合成
[0149][0150]
将20(7mg)、udp-gal(80mg)溶解在4ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至7.5,最后加入适量的酶nmlgtb。加入双蒸水至终体积为3.5ml。将该反应体系至于恒温摇床中以225r/min转速,37℃反应2h。过半制备型液相得白色固体24(4.4mg,产率:56%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.539min,esi-ms:m/z:[m h]
calcd for c
60h98n11o33
1500.63,found:1499.74;[m 2h]
2
calcd for c
60h99n11o332
750.82,found:750.57;[m nh4]
2
calcd for c
60h101n12o332
758.83,found:758.72。
[0151]
化合物25的合成
[0152][0153]
将24(2mg)、cmp-neu5ac(10.6mg)溶解在4ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至8.5,最后加入酶pmst1 m144d。加入双蒸水至终体积为1ml。将该反应体系至于恒温摇床中以225r/min转速,37℃反应4h。过半制备型液相得白色固体25(1.3mg,产率:54%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent c:25mm nh4oac;solvent d:ch3cn containing 10%25mm nh4oac,5-95%d in c over 15min,λ=220nm,保留时间为6.410min,esi-ms:m/z:[m h]
calcd for c
71h115n12o41
1791.84,found:1791.84;[m 2h]
2
calcd for c
71h16n12o412
896.37,found:895.94。
[0154]
化合物26的合成
[0155][0156]
将21(3mg)、udp-gal(59.6mg)溶解在4ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至7.5,最后加入酶nmlgtb。加入双蒸水至终体积为1.5ml。将该反应体系至于恒温摇床中以225r/min转速,37℃反应3.5h。过半制备型液相得白色固体26(1.9mg,产率:52%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent a:h2o 0.1%tfa;solvent b:ch3cn,5-95%b in a over 15min,λ=220nm,保留时间为6.271min,esi-ms:m/z:[m h]
calcd for c
74h121n12o43
1865.76,found:1864.66;[m 2h]
2
calcd for c
74h122n12o432
933.39,found:932.90;[m nh4]
2
calcd for c
74h124n13o432
941.40,found:941.38。
[0157]
化合物27的合成
[0158][0159]
将26(1.3mg,0.0002681mmol,1.0eq)、cmp-neu5ac(11.2mg)溶解在4ml离心管中,加入mgcl2(终浓度为20mmol/l),加入tris-hcl缓冲溶液使其终浓度为100mmol/l,调节ph至8.5,最后加入酶pmst1 m144d 2mg。加入双蒸水至终体积为0.75ml。将该反应体系至于恒温摇床中以225r/min转速,37℃反应15.5h。过半制备型液相得白色固体27(0.8mg,产率:47%)。通过hplc和esi-ms鉴定,hplc色谱条件:solvent c:25mm nh4oac;solvent d:ch3cn containing 10%25mm nh4oac,5-95%d in c over 15min,λ=220nm,保留时间为6.083min,esi-ms:m/z:[m 2h]
2
calcd for c
96h156n14o592
1224.98,found:1223.88;[m 2na]
2
calcd for c
96h154n14
na2o
592
1246.96,found:1246.22,[m na]
2
calcd for c
96h154n14
nao
592
1235.47,found:1235.04,[m k]
2
calcd for c
96h154
kn
14
nao
593
828.97,found:829.14。
[0160]
最后应该说明的是,以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
