1.本发明涉及药物化学制备领域,具体涉及一种治疗癫痫的药物的制备方法。
背景技术:
2.一种治疗癫痫的药物,于2008年9月获欧盟批准上市,是德国schwarz biosciences公司研发的治疗癫痫和神经性疼痛的药物,是一种新型n-甲基-d-门冬氨酸(nmda)受体甘氨酸位点拮抗剂,属于新一类功能性氨基酸,是具有双重机制的抗惊厥药。
3.目前治疗癫痫的药物的合成路线包括:
4.(1)以丝氨酸为起始原料,先与苄胺进行酰胺化,后进行羧基的甲基化,再进行胺基的乙酰化。
5.这条路线不仅使用昂贵的化学试剂,而且副产物多,产物难以纯化,不具有规模化应用价值。
6.(2)以n-boc-d-丝氨酸或者n-cbz-d-丝氨酸为原料,先进行o-甲基化作用,再与苄胺形成酰胺,再经脱保护、n-乙酰化合成治疗癫痫的药物。
7.例如,国家知识产权局于2007年06月27日公开了公开号为cn1989102a,名称为改进的拉科酰胺的合成方法的专利,该专利的说明书中公开了两种o-甲基化的方法:
8.方法a:在氮气环境下,将n-boc-d-丝氨酸在无水四氢呋喃中的溶液冷却至《-10℃。向干漏斗中加入15%w/w的正丁基锂/己烷,同时保持温度《10℃。将生成的浆液在0~5℃下老化1小时。在保持0~5℃的温度的同时,添加硫酸二甲酯,并将反应混合物在0~5℃下老化9小时。通过添加水,用30%氢氧化钠碱化至ph10~13来完成反应,并在真空下蒸发四氢呋喃/己烷。将残余物用甲苯洗涤,然后用50%柠檬酸酸化至ph<3.5。将酸化的水相用二氯甲烷萃取并且合并的萃取物经共沸蒸馏干燥。蒸发后的产量为23.7g,100%。hplc纯度为90.0%,手性纯度为100%。
9.方法b:将n-boc-d-丝氨酸和四丁基溴化铵在甲苯中的悬浮液冷却至《10℃。20%氢氧化钠在保持温度<10℃的同时向其中加入,所得混合物在<10℃下老化30分钟。在保持温度<10℃的同时添加硫酸二甲酯和50%氢氧化钠,并将反应混合物在10℃下老化1小时。向该混合物中加入水,并分离各相。用50%柠檬酸将水层酸化至ph<3.5,用二氯甲烷萃取,合并的萃取液通过共沸蒸馏干燥。蒸发后的产率为27.5g,100%,hplc纯度为96.3%,手性纯度为98.1%。
10.上述路线存在以下问题:
11.第一,需要用到大量硫酸二甲酯及昂贵的相转移催化剂四丁基溴化铵或者正丁基锂,工业化生产成本较高;
12.第二,o-甲基化过程需要控制温度在10℃以下,能源损耗较大;
13.第三,第二步以二氯甲烷为溶剂,回收过程损耗较大,环境污染较大;
14.第四,后处理过程中用柠檬酸调节ph值,会产生大量副产物柠檬酸钠,回收困难,不符合目前提倡的“绿色合成”路线。
15.文献1(j.org.chem.1979,44,13,2299
–
2300)公开了n-boc-d-丝氨酸在无机碱(乙醇钠,异丙醇钠)作用下,在四氢呋喃中与碘甲烷进行甲基化反应。此方法收率仅为50%,且有20%的外消旋产物,也不适合进行大规模工业化生产。
16.针对o-甲基化的条件,现有技术进行了多种优化,例如:
17.国家知识产权局于2009.12.02公开了申请号为200910058381.8,名称为“合成治疗拉考沙胺的新方法”的发明专利,公开的o-甲基化条件为:起始物料为n-boc-d-丝氨酸,溶剂为thf,碱为nah,甲基化试剂为ch3i。ch3i试剂昂贵,nah使用过程比较危险,且产物消旋比较严重。
18.国家知识产权局于2014.09.10公开了申请号为201410091163.5,名称为“一种拉科酰胺的制备方法”的发明专利,公开了o-甲基化条件为:起始物料为n-boc-d-丝氨酸,不使用溶剂,碱为naoh,甲基化试剂为硫酸二甲酯。在碱的水溶液中甲基化,甲基化试剂用量高,反应体系不易搅拌,收率不稳定。
19.国家知识产权局于2017.05.24公开了申请号为201510431541.4,名称为“一种拉科酰胺中间体的甲基化方法”的发明专利,公开了o-甲基化条件为:起始物料为n-boc-d-丝氨酸,溶剂为thf,碱为naoh,甲基化试剂为甲磺酸甲酯和对甲苯磺酸甲酯。利用相转移催化剂,在有水体系下甲基化试剂用量较高。
20.国家知识产权局于2020.06.12公开了申请号为201811476754.9,名称为“一种拉考沙胺的制备方法”的发明专利,公开了o-甲基化条件为:起始物料为n-boc-d-丝氨酸,溶剂为thf,碱为naoh,甲基化试剂为三甲基氧鎓四氟硼酸盐。在有水体系下甲基化试剂用量较高,且甲基化试剂较贵。
技术实现要素:
21.针对上述缺陷,本技术提供了一种全新的治疗癫痫的药物合成路线。该合成路线不再使用相转移催化试剂和正丁基锂,无水条件下,直接使用无机碱与甲基化试剂在溶剂中完成o-甲基化,甲基化试剂用量少,基本不产生外消旋产物,且具有较高的收率。
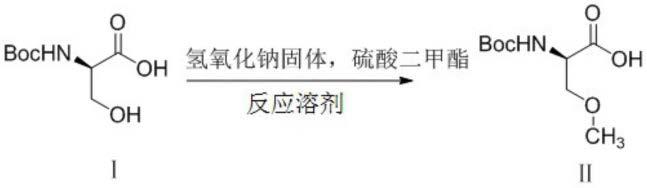
22.本发明目的之一是提供另一n-保护的d-丝氨酸的o-甲基化方法,具体过程如下:
[0023][0024]
在无机碱的反应溶剂中使用硫酸二甲酯进行o-甲基化,得到至少98%对映体纯度的r-对映体化合物ⅱ。
[0025]
优选地,反应温度为15℃~25℃。
[0026]
优选地,反应溶剂为thf,dcm,dmf,acn;更优选地,反应溶剂为thf。
[0027]
优选地,n-boc-d-丝氨酸:硫酸二甲酯的投料比为1:(1.1~1.5)。
[0028]
优选地,n-boc-d-丝氨酸:氢氧化钠的投料比为1:(2.1~3)。
[0029]
优选地,反应时间为3~5小时。
[0030]
优选地,后处理萃取溶剂为乙酸乙酯。
[0031]
优选地,n-boc-d-丝氨酸:乙酸乙酯投料比1:10。
[0032]
优选地,萃取液干燥后直接下一步缩合反应。
[0033]
本发明目的之二是提供了一种治疗癫痫的药物的制备方法,具体过程如下:
[0034][0035]
将上述制得的化合物ⅱ与氯甲酸异丁酯形成混合酸酐,加入缚酸剂;次混合酸酐再与苄胺在乙酸乙酯与四氢呋喃溶剂中发生缩合反应,制得(r)-2-((叔丁氧基)羰基氨基)-n-苄基-3-甲氧基丙酰胺,即化合物ⅲ。
[0036]
优选地,反应在无水和-20~25℃条件下进行;更优选地,在无水和-10~5℃条件下进行。
[0037]
优选地,化合物ⅱ:氯甲酸异丁酯的投料比为1:(1~1.5)。
[0038]
优选地,缚酸剂选自n-甲基吗啉或者三乙胺;更优选地,缚酸剂选自n-甲基吗啉。
[0039]
优选地,化合物ⅱ:n-甲基吗啉的投料比为1:(1.1~1.5)。
[0040]
优选地,化合物ⅱ:苄胺的投料比为1:(1.1~1.5)。
[0041]
优选地,缩合反应时间为3~5小时。
[0042]
将化合物ⅲ溶于丙酮,缓慢滴加浓盐酸并将该混合物老化1小时。浓缩有机相,在《20℃下该水相用30%氢氧化钠调ph10~12。水层用有机相萃取,获得(r)-2-氨基-n-苄基-3-甲氧基丙酰胺的乙酸乙酯溶液,即化合物ⅳ。
[0043]
优选地,反应在0~35℃条件下进行;更优选地,在25℃条件下进行。
[0044]
优选地,化合物ⅲ:浓盐酸的投料比为1:(2~5.5v)。
[0045]
优选地,化合物ⅲ:丙酮的投料比为1:(1~5v)。
[0046]
优选地,萃取溶剂为dcm,乙酸乙酯,乙酸异丙酯;更优选地,萃取溶剂为乙酸乙酯。
[0047]
优选地,水解反应时间为3~5小时。
[0048]
将化合物ⅳ的乙酸乙酯溶液冷却至《10℃并在《15℃下加入乙酸酐,加毕,混合液中加无机碱反应2小时。该混合物用水洗涤。有机相干燥,体系过滤后加正庚烷重结晶,离心分离出得到治疗癫痫的药物,即化合物v。
[0049]
优选地,反应在0~35℃条件下进行;更优选地,在20℃条件下进行。
[0050]
优选地,化合物ⅳ:乙酸酐的投料比为1:(1~2)。
[0051]
优选地,化合物ⅳ:无机碱的投料比为1:(1~2)。
[0052]
优选地,无机碱为碳酸钠,碳酸钾,碳酸氢钠,氢氧化钠,氢氧化钾;更优选地,无机碱为碳酸氢钠。
[0053]
优选地,化合物ⅳ:乙酸乙酯的投料比为1:(5~30v);更优选地,化合物ⅳ:溶剂的投料比为1:15v。
[0054]
优选地,化合物ⅳ:正庚烷的投料比为1:(5~30v);更优选地,化合物ⅳ:溶剂的投料比为1:15v。
[0055]
优选地,化合物重结晶温度为-15~25℃;更优选地,在0℃条件结晶最优。
[0056]
本发明的有益效果为:1、本发明的方法中d-丝氨酸的o-甲基化期间,基本上没有形成甲基酯并且产物没有显著是外消旋化,导致本法制备治疗癫痫的药物产率提高并且产物对映体纯度的提高,总收率80%左右,手性纯度99.95%。
[0057]
2、在与技术背景描述的路线相比,减少了硫酸二甲酯的用量,且反应在温和条件下进行,减少了能源消耗,降低成本;使用低毒的乙酸乙酯做溶剂,使得烷基化后的产物不用柠檬酸处理,减少了副产物的环境污染,使得本发明更符合低毒,低污染的绿色合成要求。
具体实施方式
[0058]
以下将结合实施例对本发明作进一步的详细描述,本发明的实施例仅用于说明本发明的技术方案,并非对本发明的限制,凡依照本发明公开的内容所作的任何本领域的等同置换,均属于本发明的保护范围。
[0059]
本公开所用化学试剂可来自商业途径。
[0060]
本公开中有关物质含量或纯度可通过hplc方法检测获得:
[0061]
色谱条件:
[0062]
色谱柱:inertsil ods-3,250mm
×
4.6mm
×
5μm;
[0063]
检测波长:205nm、230nm;
[0064]
柱温:30℃;
[0065]
流速:1.2ml/min;
[0066]
流动相:乙腈/磷酸二氢钾缓冲液,梯度洗脱。
[0067]
化合物的结构通过核磁共振(1hnmr)来确定的。
[0068]
核磁共振(1hnmr)位移(δ)以百万分之一(ppm)的单位给出;核磁共振(1hnmr)的测定是用brukeravance-300核磁仪,测定溶剂为氘代氯仿(cdcl3-d6),内标为四甲基硅烷(tms),化学位移是以10-6(ppm)作为单位给出。
[0069]
氮气是指反应瓶连接一个1l容积的氮气气球。
[0070]
本发明的术语“室温”是指温度处于10℃~25℃之间。
[0071]
实施例1化合物ⅱ,(r)-2-n-boc-氨基-3-甲氧基丙酸的合成
[0072]
在氮气保护下,将n-boc-d-丝氨酸(40g,0.195mol)四氢呋喃(400ml)溶液冷却至15℃~25℃。向其中加固体氢氧化钠(23g,0.585mol)搅拌老化半小时,同时保持温度在15℃~25℃。加入硫酸二甲酯(36.9g,0.293mol),同时保持温度15℃~25℃,反应混合物在20℃搅拌4小时。混合物体系降温至10℃以下,用3nhcl酸化至ph《3.5,用400ml乙酸乙酯萃取,有机相用饱和食盐水(100ml*2)洗涤,有机相硫酸钠干燥后过滤得到萃取物。蒸发后42.7g,收率100%,手性纯度:99%以上。
[0073]
实施例2化合物ⅱ,(r)-2-n-boc-氨基-3-甲氧基丙酸的合成
[0074]
在氮气保护下,将n-boc-d-丝氨酸(40g,0.195mol)四氢呋喃(400ml)溶液冷却至15℃~25℃。向其中加固体氢氧化钾(32.8g,0.585mol)搅拌老化半小时,同时保持温度15
℃~25℃。加入硫酸二甲酯(36.9g,0.293mol),同时保持温度15℃~25℃,反应混合物在20℃搅拌4小时。混合物体系降温至10℃以下,用3nhcl酸化至ph《3.5,用400ml乙酸乙酯萃取,有机相用饱和食盐水(100ml*2)洗涤,有机相硫酸钠干燥后过滤得到萃取物。蒸发后42.2g,收率99%。
[0075]
实施例3化合物ⅱ,(r)-2-n-boc-氨基-3-甲氧基丙酸的合成
[0076]
在氮气保护下,将n-boc-d-丝氨酸(40g,0.195mol)乙腈(400ml)溶液冷却至15℃~25℃。向其中加固体氢氧化钠(23g,0.585mol)搅拌老化半小时,同时保持温度15℃~25℃。加入硫酸二甲酯(36.9g,0.293mol),同时保持温度15℃~25℃,反应混合物在20℃搅拌4小时。混合物体系降温至10℃以下,用3nhcl酸化至ph《3.5,用400ml乙酸乙酯萃取,有机相用饱水(100ml*3)洗涤,有机相硫酸钠干燥后浓缩干得到(r)-2-n-boc-氨基-3-甲氧基丙酸,蒸发后42.2g,收率99%。
[0077]
实施例4化合物ⅱ,(r)-2-n-boc-氨基-3-甲氧基丙酸的合成
[0078]
在氮气保护下,将n-boc-d-丝氨酸(40g,0.195mol)二氯甲烷(400ml)溶液冷却至15℃~25℃。向其中加固体氢氧化钠(23g,0.585mol)搅拌老化半小时,同时保持温度15℃~25℃。加入硫酸二甲酯(36.9g,0.293mol),同时保持温度15℃~25℃,反应混合物在20℃搅拌4小时。混合物体系降温至10℃以下,加50ml水,用3nhcl酸化至ph.3-4,分液,有机相用饱和氯化钠(100ml*2)洗涤,有机相硫酸钠干燥后浓缩干得到(r)-2-n-boc-氨基-3-甲氧基丙酸,蒸发后40.6g,收率95%。
[0079]
实施例5化合物ⅲ,(r)-2-((叔丁氧基)羰基氨基)-n-苄基-3-甲氧基丙酰胺的合成
[0080]
将实施例1制得的萃取溶液冷却至《-10℃并在《-5℃加入氯甲酸异丁酯(26.6g,0.195mol)。在《-5℃下加n-甲基吗啉(29.5g,0.293mol)并在-10
±
5℃下反应1小时。在《-5℃下加入苄胺(25g,0.234mol)并使体系恢复室温搅拌2小时。体系用水(100ml),1nhcl(100ml),饱和碳酸氢钠(100ml)和水(100ml)洗涤该混合物,体系干燥浓缩后用100ml正庚烷打浆,离心干燥得到化合物ⅲ55g,以化合物ⅱ记收率91.6%,手性纯度:99.9%。
[0081]
实施例6化合物ⅳ,(r)-2-氨基-n-苄基-3-甲氧基丙酰胺的合成
[0082]
将化合物ⅲ(40g,0.130mol)溶于40ml丙酮,缓慢滴加浓盐酸(46.5ml,0.541mol)并将该混合物老化1小时。体系浓缩掉有机相,在《20℃下该水相用30%氢氧化钠调ph10-12。水层用乙酸乙酯(3*200ml)萃取,合并有机层并用水(200ml)洗涤,获得(r)-2-氨基-n-苄基-3-甲氧基丙酰胺的乙酸乙酯溶液。蒸发后36.1g,收率99%。
[0083]
实施例7治疗癫痫的药物,can-2-(乙酰基氨基)-n-苯甲基-3-甲氧基丙酰胺的合成
[0084]
将如上制得的(r)-2-氨基-n-苄基-3-甲氧基丙酰胺的乙酸乙酯溶液冷却至《10℃和并在《15℃下加入乙酸酐(13.8g,0.130mol),加毕,混合液中加碳酸氢钠(18g,0.2mol)反应2小时。该混合物用水(2*50ml)洗涤,体系干燥过滤后加热至50℃,加500ml正庚烷搅拌。体系缓慢冷却至0-5℃结晶,离心分离出治疗癫痫的药物30.8g,收率90%,hplc纯度99.95%,手性纯度:99.95%。
[0085]
1h nmr(400mhz,dmso)δ8.49(d,j=6.0hz,2h),8.10(d,j=8.0hz,1h),7.34
–
7.28(m,1h),7.27
–
7.20(m,2h),4.5(m,1h),4.29(d,2h),3.51(qd,j=9.7,5.8hz,2h),3.25(s,
3h),1.88(s,3h).13c nmr(101mhz,dmso)δ169.7,169.4,139.3,128.2,127.1,126.0,71.3,56.6,52.6,43.2,18.4.
[0086]
试验例1
[0087]
o-甲基化的溶剂、碱、甲基化试剂筛选试验:
[0088]
溶剂选择
[0089][0090]
上表是溶剂对中间体ii收率的影响,其中甲基化试剂为硫酸二甲酯,碱为氢氧化钠。乙腈和二氯甲烷做溶剂收率偏低,原因在于二甲基杂质的不可避免的产生,所以最终溶剂选择为四氢呋喃。
[0091]
碱的选择
[0092][0093]
上表是碱对中间体ii收率的影响,其中甲基化试剂为硫酸二甲酯,溶剂为四氢呋喃。氢氧化钾与氢氧化钠收率相当,碳酸钾的收率明显降低,考虑到成本选择氢氧化钠。
[0094]
甲基化试剂的选择
[0095][0096]
上表是甲基化试剂对中间体ii收率的影响,其中碱为氢氧化钠,溶剂为四氢呋喃。其中碳酸二甲酯的收率明显降低,碘甲烷与硫酸二甲酯收率相差不大,考虑到碘甲烷毒性更大,成本高,甲基化试剂选择硫酸二甲酯。
[0097]
对比例1:与专利200910058381.8的对比
[0098]
本技术与专利200910058381.8的区别在于碱和甲基化试剂不同,如下表所示:
[0099]
专利碱甲基化试剂反应是否无水甲基化试剂用量200910058381.8nahch3i是1.1eq本技术naoh硫酸二甲酯是1.5eq
[0100]
对比例2:与专利201410091163.5的对比
[0101]
本技术与专利201410091163.5的区别在于是否使用有机溶剂,如下表所示:
[0102][0103]
对比例3:与专利201510431541.4的对比
[0104]
本技术与专利201510431541.4的区别在于甲基化试剂和反应温度,如下表所示:
[0105][0106]
对比例4:与专利201811476754.9的对比
[0107]
本技术与专利201811476754.9的区别在于甲基化试剂不同,如下表所示:
[0108]
专利甲基化试剂反应是否无水甲基化试剂用量相转移催化剂201811476754.9三甲基氧鎓四氟硼酸盐否3eq有本技术硫酸二甲酯是1.5eq无
[0109]
上述实施例仅仅是为清楚地说明本发明创造所作的举例,而并非对本发明创造具体实施方式的限定。对于所属领域的技术人员来说,在上述说明的基础上还可以做出其它不同形式的修改或改进。由此所引申出的显而易见的修改或改进仍处于本发明创造权利要求的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。