一种发光增强水相检测bi
3
的发光晶体材料的制备方法
技术领域
1.本发明属于发光检测晶体材料技术领域,具体涉及一种发光增强水相检测 bi
3
的发光晶体材料的制备方法。
背景技术:
2.三价金属铋离子(bi
3
)广泛应用在制药行业,例如用于制作抗菌剂、抗艾 滋病毒剂、抗溃疡剂和放射治疗剂等。然而,过量的bi
3
将对人类健康和生态环 境造成威胁。土壤中bi
3
浓度超过0.416g/kg,将会影响蚯蚓的繁殖率和数量, 进而影响土壤通气和土壤的疏松度。并且bi
3
半衰期比较长,可能在肾脏中积累 导致肾病。因此,探索高效检测bi
3
的材料显得极为重要。
3.现有的检测bi
3
的发光材料很少,具有灵敏的发光增强水相检测bi
3
的发光 晶体材料还未报道。因此开发灵敏的发光增强水相检测bi
3
的发光晶体材料的制 备方法和产品具有重要意义。
技术实现要素:
4.本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳 实施例。在本部分以及本技术的说明书摘要和发明名称中可能会做些简化或省 略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能 用于限制本发明的范围。
5.鉴于上述及现有技术中存在的问题,提出了本发明。
6.因此,本发明的目的在于提供一种发光增强水相检测bi
3
的发光晶体材 料的制备方法。
7.为解决上述技术问题,根据本发明的一个方面,本发明提供了如下技术方案: 一种发光增强水相检测bi
3
的发光晶体材料的制备方法,包括,
8.将氯化镉、1,4-萘二甲酸(h2nda)和9-二(3-吡啶)乙烯-芴(3-l)加入到 混合溶剂中,搅拌制得混合液;
9.将混合液置于密闭的反应釜中加热反应,然后缓慢降至室温,产物经过滤、 超声洗涤、干燥得到发光晶体材料[cd(nda)(3-l)(h2o)]n。
[0010]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选方案,其中:所述氯化镉、1,4-萘二甲酸(h2nda)和9-二(3-吡啶)乙烯
‑ꢀ
芴(3-l)的摩尔比为1:1:0.25~0.75。
[0011]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选方案,其中:所述混合溶剂为水与乙腈体积比为1:0.5~2的混合溶液。
[0012]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选方案,其中:所述每添加0.1mmol氯化镉所需混合溶剂的体积为4~8ml。
[0013]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选
方案,其中:所述加热反应,加热温度为130~150℃,加热时间为 24~72h。
[0014]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选方案,其中:所述降至室温的降温速率为2~5℃/h。
[0015]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选方案,其中:所述超声洗涤,洗涤溶剂为水和乙腈体积比为1:0.5~2混 合物,超声功率为50hz,超声时间为5~10min。。
[0016]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法的一 种优选方案,其中:所述干燥,包括真空干燥或室温干燥,干燥温度50~80℃, 干燥时间3~10min。
[0017]
作为本发明所述发光增强水相检测bi
3
的发光晶体材料的制备方法制备 所得的材料[cd(nda)(3-l)(h2o)]n,其中,n无限制范围。
[0018]
作为本发明所述材料[cd(nda)(3-l)(h2o)]n的一种优选方案,其中:所述材 料[cd(nda)(3-l)(h2o)]n检测溶液中bi
3
的增强常数为-5.3
×
104m-1
,检测限为1.87
ꢀ×
10-5
mm。
[0019]
本发明的有益效果:
[0020]
本发明提供了一种发光增强水相检测bi
3
的发光晶体材料[cd(nda)(3
‑ꢀ
l)(h2o)]n的制备方法和产品,将氯化镉、1,4-萘二甲酸和9-二(3-吡啶)乙烯-芴加 入到水和乙腈混合液中,搅拌制得混合液,将该混合液置于密闭的反应釜中加热 反应,然后缓慢降至室温,产物经过滤、洗涤、干燥即得到发光晶体材料。本发 明首次获得了一种高灵敏发光增强水相检测bi
3
的发光晶体材料[cd(nda)(3
‑ꢀ
l)(h2o)]n,合成路线简单易控,适合工业化生成,制备所得材料[cd(nda)(3
‑ꢀ
l)(h2o)]n产率最高可达60%。增强常数k
sv
=-5.3
×
104m-1
,检测限为1.87
×
10-5 mm。
附图说明
[0021]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需 要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一 些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还 可以根据这些附图获得其它的附图。其中:
[0022]
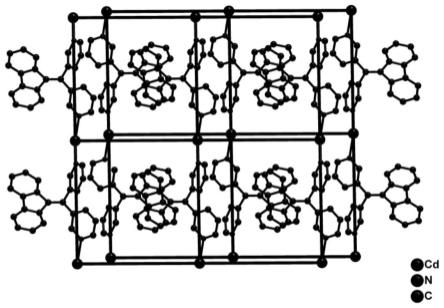
图1为晶体材料[cd(nda)(3-l)(h2o)]n的二维晶体结构图(氢原子被省略);
[0023]
图2为所制备的晶体材料[cd(nda)(3-l)(h2o)]n的粉末x-射线衍射谱图;
[0024]
图3为2.5ml晶体材料[cd(nda)(3-l)(h2o)]n的水悬浮液(0.2g/ml)的激发 发射图谱;
[0025]
图4为2.5ml晶体材料[cd(nda)(3-l)(h2o)]n的水悬浮液(0.2mg/ml)中 加入不同体积bi
3
水溶液(2mmol/l)的发光强度变化曲线图;
[0026]
图5为晶体材料[cd(nda)(3-l)(h2o)]n检测bi
3
的增强常数曲线;图6为晶体材料[cd(nda)(3-l)(h2o)]n检测bi
3
的检测限曲线。
具体实施方式
[0027]
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实 施例对本发明的具体实施方式做详细的说明。
[0028]
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明 还可
以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不 违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例 的限制。
[0029]
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一 个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个 实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相 排斥的实施例。
[0030]
本发明实施例中所用化学试剂,若无特殊说明,均为普通市售分析纯。
[0031]
本发明实施例中产率的计算方法为:基于cd计算产率,计算步骤为:
[0032][0033]
实施例中所使用9-二(3-吡啶)乙烯-芴为实验室自制,具体制备过程如下:
[0034]
将9-芴酮,四溴化碳和三苯基膦加入无水二氯甲烷中,在氮气保护下加热 至30~60℃,反应12~36小时,反应结束后冷却,采用柱层析法,以正己烷为洗 脱剂,得到粗产物,最后用正己烷重结晶得到黄色产物9-二溴乙烯-芴;
[0035]
将9-二溴乙烯-芴,3-吡啶硼酸,无水碳酸钠,三苯基膦和醋酸钯催化剂加 入1,4-二氧六环与水的混合溶液中,在氮气保护下加热至100~140℃,反应10~20 小时,反应结束后,用乙酸乙酯萃取3遍,收集有机层,用无水硫酸钠干燥,减 压旋蒸,采用柱层析法,以乙酸乙酯:石油醚(v:v=1:1)为洗脱剂得到粗产物, 再用乙酸乙酯洗涤得到淡黄色产物9-二(3-吡啶)乙烯-芴。
[0036]
实施例1:
[0037]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.025mmol 9-二(3-吡啶)乙烯
ꢀ‑
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0038]
将制得的混合液置于密闭的反应釜中加热至140℃反应72h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液超声洗涤、烘箱60℃干 燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:40%。
[0039]
实施例2:
[0040]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.05mmol 9-二(3-吡啶)乙烯
‑ꢀ
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0041]
将制得的混合液置于密闭的反应釜中加热至140℃反应72h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液50hz超声洗涤10min、 烘箱60℃干燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:60%。
[0042]
实施例3:
[0043]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.075mmol 9-二(3-吡啶)乙烯
ꢀ‑
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0044]
将制得的混合液置于密闭的反应釜中加热至140℃反应72h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液50hz超声洗涤10min、 烘箱60℃干燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:55%。
[0045]
实施例4:
[0046]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.05mmol 9-二(3-吡啶)乙烯
‑ꢀ
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0047]
将制得的混合液置于密闭的反应釜中加热至140℃反应24h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液50hz超声洗涤10min、 烘箱60℃干燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:40%。
[0048]
实施例5:
[0049]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.05mmol 9-二(3-吡啶)乙烯
‑ꢀ
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0050]
将制得的混合液置于密闭的反应釜中加热至140℃反应48h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液50hz超声洗涤10min、 烘箱60℃干燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:45%。
[0051]
实施例6:
[0052]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.05mmol 9-二(3-吡啶)乙烯
‑ꢀ
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0053]
将制得的混合液置于密闭的反应釜中加热至130℃反应72h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液50hz超声洗涤10min、 烘箱60℃干燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:50%。
[0054]
实施例7:
[0055]
将0.1mmol氯化镉、0.1mmol 1,4-萘二甲酸和0.05mmol 9-二(3-吡啶)乙烯
‑ꢀ
芴加入到6ml的水和乙腈(v:v=1:1)中搅拌制得混合液;
[0056]
将制得的混合液置于密闭的反应釜中加热至150℃反应72h,以5℃/h的 速率缓慢降温到室温后,经过滤、用乙腈和水混合溶液50hz超声洗涤10min、 烘箱60℃干燥5min,即得到[cd(nda)(3-l)(h2o)]n晶体材料。计算产率为:30%。
[0057]
bi
3
探测性能检测:取5mg制备的晶体材料[cd(nda)(3-l)(h2o)]n分散到25 ml的水中制得稳定的悬浮液,向悬浮液中加入不同体积含bi
3
的水溶液(2 mmol/l),并分别在320nm的激发光下测试其荧光强度。
[0058]
本发明所制备的晶体材料[cd(nda)(3-l)(h2o)]n的粉末x-射线衍射衍射图样 与理论计算的x-射线衍射图样基本一致,说明本发明所制备的晶体材料具有很 高的纯度。浸泡在bi
3
水溶液24h后的粉末x-射线衍射图样与理论计算的x-射 线衍射图样基本一致,说明该晶体材料在bi
3
水溶液中具有很好的稳定性。
[0059]
图4为2.5ml晶体材料[cd(nda)(3-l)(h2o)]n的水悬浮液(0.2mg/ml)中 加入不同体积bi
3
水溶液(2mmol/l)的发光强度变化曲线图。在320nm的激发 波长下测定晶体材料的水悬浮液的荧光强度,然后向其中逐渐滴加2mmol/l的 bi
3
水溶液,随着bi
3
量的逐渐增多,其悬浮液发光强度逐渐增强。
[0060]
这可能有利于在bi
3
和芴环之间形成π-阳离子-π填充模型,从而有助于框 架内的电子跃迁,增加发射强度。随着bi
3
浓度的增加,在该框架内产生的π-阳 离子-π填充模型将不断增加,有利于提高选择效率增加,并相应导致发射强度 的增加。
[0061]
本发明提供了一种发光增强水相检测bi
3
的发光晶体材料[cd(nda)(3
‑ꢀ
l)(h2o)]n的制备方法和产品,将氯化镉、1,4-萘二甲酸和9-二(3-吡啶)乙烯-芴加 入到水和乙腈混合液中,搅拌制得混合液,将该混合液置于密闭的反应釜中加热 反应,然后缓慢降至室温,产物经过滤、洗涤、干燥即得到发光晶体材料。本发 明首次获得了一种高灵敏发光
增强水相检测bi
3
的发光晶体材料[cd(nda)(3
‑ꢀ
l)(h2o)]n,合成路线简单易控,适合工业化生成,制备所得材料[cd(nda)(3
‑ꢀ
l)(h2o)]n产率最高可达60%。,增强常数k
sv
=-5.3
×
104m-1
,检测限为1.87
×ꢀ
10-5
mm。
[0062]
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照 较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对 本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和 范围,其均应涵盖在本发明的权利要求范围当中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。