手性binol三氟甲磺酸酯衍生物及其制备方法和应用
技术领域
1.本发明属于手性催化剂关键中间体的制备以及应用领域,手性binol三氟甲磺酸酯衍生物及其制备方法和应用。
背景技术:
2.自从2004年terada和akiyama课题组分别独立报道手性磷酸催化的不对称mannich反应以来,经过近20年的发展,基于binol及h8-binol的手性磷酸已经成为手性有机酸催化研究领域的优势催化剂。特别地,当binol及h8-binol骨架的3,3
′
位引入含2,6-二异丙基-4-芳基(或大位阻烷基)-苯基取代基时,在相应的不对称催化反应中往往能够取得较好的立体选择性。其中,当芳基侧链取代基r=ipr(即trip手性磷酸),9-anthryl(文中简写为9-an),及1-adamantyl(文中简写为1-ad)时的手性磷酸衍生物是这类催化剂的重要代表(图一),因此研究其高效及实用性的合成方法不仅在不对称催化领域具有学术研究价值而且在精细手性有机化学品规模化合成方面也具有潜在的应用价值。
3.目前为止,这类催化剂的合成方法都是以先获得相应芳基侧链的溴化物(或碘化物),然后将其预先制备成格氏试剂,接着利用kumada偶联将其组装至binol或者h8-binol的3,3
′‑
位。虽然这种方法对于合成r=ipr以及1-ad时不存在着问题,但当r=9-an时则存在着以下问题(x.cheng,and b.list et al.,angew.chem.int.ed.,2008,47,5079):
4.(1)r=9-an时,相应的芳基溴化物在醚类溶剂(四氢呋喃及乙醚)中溶解度很差,在制备格氏试剂时需要大量的醚类溶剂去溶解该芳基侧链;
5.(2)制备好的格氏试剂在进行kumada偶联时反应的重现性很差,经常遇到偶联反应无法发生的问题。
6.为了解决含r=9-an取代基的芳基侧链在进行kumada偶联时重现性很差的问题,list课题组最近利用negishi偶联代替kumada偶联,成功地制备了该类催化剂,提高了偶联反应过程中的重现性(m.duan,b.list,k.n.houk,and y.lan et al.angew.chem.int.ed.,2022,early view,doi:10.1002/anie.202113204)。
7.因此,寻找其他合适的合成方法,不仅可以实现r=9-an手性磷酸关键中间体的合成,简化该磷酸催化剂的合成路线;而且通过利用相同的手性中间体,高效率发散式地合成r为不同取代基的系列手性磷酸催化剂可达到加速优化催化反应条件,快速获得取代基结构与反应立体选择性之间的关系,是重要的现实问题。
技术实现要素:
8.本技术的目的在于提供一种手性binol三氟甲磺酸酯衍生物及其制备方法和应用,旨在解决以下合成大位阻手性磷酸时遇到的技术问题:
9.(1)在往binol或h8-binol的3,3
’‑
位安装含2,6-二异丙基-4-芳基(或大位阻烷基)-苯基烷基侧链时,现存的方法需要一一对应地合成相关中间体,不仅耗时耗力,而且无法快速获得这类催化剂芳基侧链4-位取代基位阻、电子性质与催化反应立体选择性之间的
关系;
10.(2)简化侧链2,6-二异丙基-4-芳基苯基,芳基为9-an取代基时手性磷酸的合成难度,提高合成的重现性。
11.(3)可发散式合成不同r(r为芳基)取代的binol衍生物(通式见iii)。
12.本发明提供的方法不仅在基础研究上具有重要的参考价值,而且本法提供的合成方法所合成的中间体与手性催化剂均具有一定的商品化价值。
13.本发明提供的手性binol三氟甲磺酸酯衍生物为式i所示的化合物、或其对映体、消旋体中的至少一种:
[0014][0015]
式中:ipr为异丙基,otf为三氟甲磺酸酯基。
[0016]
上述的手性binol三氟甲磺酸酯衍生物的制备方法,包括如下步骤:将式ii所示的苯酚衍生物与镁反应预先制备得到相应的格氏试剂,然后与手性binol或h8-binol的双卤代物进行kumada偶联反应,所获得的产物经脱pg保护基后与三氟甲磺酸酐反应而获得式i;
[0017][0018]
式中:pg选自mom、bn、三苯甲基或硅基保护基中的至少一种。
[0019]
进一步的,所述手性binol的双卤代物的结构通式如下:
[0020][0021]
式中:x选自溴或碘中的至少一种。
[0022]
进一步的,进行kumada偶联过程中所使用的催化剂为1-20mol%的ni(pph3)2cl2(ni(pph3)2cl2用量为手性binol或h8-binol的双卤代物摩尔量的1-20%)。
[0023]
本发明还提供了手性binol三氟甲磺酸酯衍生物的应用,具体的,用于与硼酸衍生物rb(oh)2经suzuki反应,合成式iii所示的大位阻手性磷酸中间体,
[0024][0025]
式中:r选自取代的芳基、烷基或聚合物中的至少一种。
[0026]
进一步的,利用suzuki偶联合成iii的过程中,使用了催化剂且该催化剂为四(三苯基膦)钯pd(pph3)4或三(二亚苄基丙酮)二钯pd2(dba)3中的至少一种。
[0027]
进一步的,suzuki偶联过程中,采用了溶剂且该溶剂为四氢呋喃、乙醚、1,4-二氧六环或乙二醇二甲醚中的至少一种或任意多种与水的任意比例的混合溶剂。
[0028]
进一步的,suzuki偶联过程中,采用了碱且该碱为碳酸钠、碳酸钾、碳酸氢钠、磷酸钠或磷酸钾中的至少一种。
[0029]
上述大位阻手性磷酸中间体用于合成式v所示的手性磷酸催化剂,
[0030][0031]
式中,x选自氧原子,硫原子,或nso2r中的任意一种,r选自氢、c1-c18的烷基或全氟烷基,或者为芳基中的至少一种。
[0032]
与现有技术相比,本发明取得了如下技术优势:本发明提供了一种3,3
′‑
位含2,6-二异丙基-4-苯酚三氟甲磺酸酯取代基的手性binol衍生物,基于该衍生物不仅可容易解决list-cheng手性磷酸催化剂中间体8合成重复性差的问题,并最终得到具有高价值的手性磷酸催化剂10(该催化剂在大赛璐的价格》3000元人民币/100mg),而且可以合成不同r取代的全新的大位阻binol衍生物,这类衍生物可容易地转化成新型手性磷酸或其衍生物。
附图说明
[0033]
图1为含2,6-二异丙基-4-r-苯基取代基手性磷酸通式及目前文献中采用的逆合成路线。
[0034]
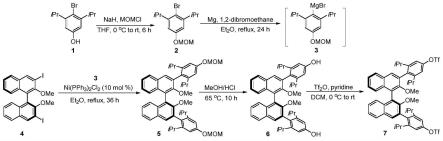
图2为本发明实施例中的手性binol三氟甲磺酸酯衍生物的合成路线。
[0035]
图3为本发明应用实施例1中芳基侧链4-位取代基为9-an的手性磷酸的合成路线。
[0036]
图4为本发明应用实施例2中大位阻手性磷酸中间体11的合成路线。
[0037]
图5为本发明应用实施例3中大位阻手性磷酸中间体12的合成路线。
[0038]
图6为本发明实施例1中中间体2的氢谱图。
[0039]
图7为本发明实施例1中中间体2的碳谱图。
[0040]
图8为本发明实施例1中中间体5的氢谱图。
[0041]
图9为本发明实施例1中中间体5的碳谱图。
[0042]
图10为本发明实施例1中中间体6的氢谱图。
[0043]
图11为本发明实施例1中中间体6的碳谱图。
[0044]
图12为本发明实施例1中中间体7的氢谱图。
[0045]
图13为本发明实施例1中中间体7的碳谱图。
[0046]
图14为本发明实施例1中中间体7的氟谱图。
[0047]
图15为本发明应用实施例1中手性磷酸10的氢谱图。
[0048]
图16为本发明应用实施例1中手性磷酸10的磷谱图。
[0049]
图17为本发明应用实施例2中手性磷酸中间体11的氢谱图。
[0050]
图18为本发明应用实施例2中手性磷酸中间体11的碳谱图。
[0051]
图19为本发明应用实施例2中手性磷酸中间体12的氢谱图。
[0052]
图20为本发明应用实施例2中手性磷酸中间体12的碳谱图。
具体实施方式
[0053]
为了使本技术要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本技术进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本技术,并不用于限定本技术。本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
[0054]
实施例1:3,3
′‑
位含2,6-二异丙基-4-苯酚三氟甲磺酸酯取代基的手性binol衍生物的合成:以binol骨架为例,但不局限于binol骨架;苯酚衍生物1酚羟基保护基pg为mom保护基为例,但不局限于该保护基。以下给出一个代表性的合成路线。
[0055][0056]
(a)mom保护的中间体2的合成:0℃于氮气保护下,将已知化合物1(2.06g,8.01mmol)溶解于超干四氢呋喃(10ml),搅拌情况下,分批缓慢加入氢化钠(480.8mg,12.02mmol,1.5equiv,60wt%in mineral oil),此时可观察到有大量的气泡冒出,在相同的反应温度下继续搅拌30分钟,然后用注射器将momcl(838.1mg,10.41mmol,1.3equiv)缓慢滴加至反应瓶中,滴加完毕后将反应体系升温至室温,并搅拌6小时。tlc显示底物1完全转化,往反应瓶中缓慢滴加水(10ml)淬灭过量的氢化钠,乙酸乙酯萃取(3*10ml),无水硫酸钠干燥,过滤,旋蒸溶剂而获得粗产物,最后经硅胶柱层析而获得产物2(无色油状物,2.15g,89%yield,淋洗剂:石油醚/乙酸乙酯=20:1)。
[0057]1h nmr(400mhz,cdcl3)δ6.85(s,2h),5.17(s,2h),3.53
–
3.46(m,5h),1.24(d,j=4.0hz,12h)ppm.
[0058]
13
c nmr(101mhz,cdcl3)δ157.0,149.1,118.5,112.4,94.8,56.2,33.7,23.0ppm.hrms(es )calcd for c
14h22
bro2[m h]
:303.0783,found:303.0746.
[0059]
(b)格氏试剂3的制备:氮气保护下,盐酸活化的镁屑(340.8mg,14.2mmol,2.0equiv)置于烘箱烘干的100毫升两口瓶中,并加入5ml超干的乙醚,并将上述合成的2(2.14g,7.1mmol,1.0equiv)中的约0.1ml样品,以及催化量的1,2-二溴乙烷(10mol%)加入到该反应瓶中,将以上混合物用电热枪温和加热来引发格氏反应。待格氏反应引发后(现象:电热枪略微加热后,即可观察到剧烈的乙醚沸腾回流现象,表明1,2-二溴乙烷引发了该格氏反应),停止电热枪加热,在保持体系微沸的情况下,将剩余原料2以及超干的乙醚(15ml)交替滴加入反应瓶中。滴加完毕后,将反应瓶置于油浴中回流24小时而得到3的浅黄色溶液。
[0060]
(c)中间体5的制备:n2保护下,将4(736mg,1.3mmol,1.0equiv)以及ni(pph3)2cl2(85.0mg,0.13mmol,10mol%)置于烘箱烘干的100毫升两口瓶中,并加入15ml超干的乙醚,然后将上述制备格氏试剂3的乙醚溶液缓慢滴加至该悬浊液中,在滴加过程中,反应体系的颜色逐渐由绿色变成黄绿色,再变成红色,最后变成红褐色。滴加完毕后,反应烧瓶置于油浴中回流36小时。反应结束后,缓慢滴加nh4cl饱和溶液淬灭反应体系,乙醚萃取(3*25ml),无水硫酸钠干燥,过滤旋干而得到粗产物,最后经硅胶柱层析(石油醚:乙酸乙酯=50:1至20:1)而获得中间体5(白色固体,750mg,77%yield)。
[0061]1h nmr(400mhz,cdcl3)δ7.86(d,j=8.0hz,2h),7.72(s,2h),7.44
–
7.40(m,2h),7.35
–
7.28(m,4h),6.94
–
6.91(m,4h),5.25(s,4h),3.57(s,6h),3.08(s,6h),2.90
–
2.77(m,4h),1.21(d,j=4.0hz,6h),1.16(d,j=4.0hz,6h),1.12(d,j=4.0hz,6h),1.07(d,j=4.0hz,6h)ppm.
[0062]
13
c nmr(101mhz,cdcl3)δ157.6,155.2,149.2,148.8,134.0,133.8,131.2,130.3,129.8,128.0,126.1,125.8,124.8,124.7,110.72,110.66,94.9,59.9,56.2,31.2,31.0,25.4,25.1,23.3,23.2ppm.
[0063]
hrms(es )calcd for c
50h59
o6[m h]
:755.4312,found:755.4304.
[0064]
(d)中间体6的制备:将中间体5(750mg,1.0mmol)溶解于氯仿-甲醇混合溶剂中(chcl3:meoh=3:2,50ml),然后将5ml浓盐酸加入到反应瓶中,然后在65℃油浴中回流10h。反应结束后,将反应体系冷却至室温,然后加入30ml水,dcm萃取(3*30ml),混合的有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋干得粗产物,最后经柱色谱层析后得到中间体6(白色固体,560mg,84%yield)。
[0065]1h nmr(400mhz,cdcl3)δ7.87(d,j=8.0hz,2h),7.71(s,2h),7.44
–
7.40(m,2h),7.34
–
7.27(m,4h),6.74
–
6.72(m,4h),4.88(s,2h),3.08(s,6h),2.89
–
2.75(m,4h),1.19(d,j=4.0hz,6h),1.15(d,j=4.0hz,6h),1.11(d,j=4.0hz,6h),1.05(d,j=4.0hz,6h)ppm.
[0066]
13
c nmr(101mhz,cdcl3)δ155.5,149.6,149.1,134.0,133.7,131.3,130.3,128.6,128.0,126.1,125.8,124.9,124.7,109.9,109.8,59.8,31.0,30.9,25.4,25.1,23.3,23.2ppm.
[0067]
hrms(es )calcd for c
46h51
o4[m h]
:667.3787,found:667.3730.
[0068]
(e)中间体7的制备:零度及n2保护下,将中间体6(560mg,0.84mmol,1.0equiv)溶解于超干的二氯甲烷(20ml),然后用微量注射器加入吡啶(132.9mg,1.68mmol,2.0equiv)。所得混合反应体系在相同的反应温度下搅拌15分钟,然后加入三氟甲磺酸酐(593mg,2.1mmol,2.5equiv),将反应体系升温至室温,并搅拌10小时。反应结束后,反应体系加入1.0m的hcl(20ml),dcm萃取(3*20ml),合并的有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋干,得粗产物,最后将硅胶柱层析(石油醚:乙酸乙酯=20:1)而得到中间体7(白色固体,540mg,70%yield)。
[0069]1h nmr(400mhz,cdcl3)δ7.89(d,j=8.0hz,2h),7.72(s,2h),7.47
–
7.43(m,2h),7.35
–
7.34(m,4h),7.12
–
7.09(m,4h),3.01(s,6h),2.93
–
2.86(m,2h),2.84
–
2.77(m,2h),1.21(d,j=4.0hz,6h),1.15(d,j=4.0hz,6h),1.12(d,j=4.0hz,6h),1.04(d,j=4.0hz,6h)ppm.
[0070]
13
c nmr(101mhz,cdcl3)δ154.6,150.9,150.2,150.1,135.8,134.1,132.3,130.7,130.2,128.1,126.6,125.6,125.1,124.7,120.5(q,1j
c-f
=251hz),117.0,115.6,59.9,31.2,31.1,25.2,24.7,23.01,22.95ppm.
[0071]
19
f nmr(376mhz,cdcl3)δ-72.89ppm.
[0072]
hrms(es )calcd for c
49h49
f6o
10
s2[m cooh]-:975.2671,found:975.2699.
[0073]
中间体7的应用
[0074]
为了使本技术要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对中间体7的应用进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本技术,并不用于限定本技术。
[0075]
具体应用实施例1:合成芳基侧链4-位取代基为9-an的手性磷酸
[0076][0077]
中间体8的合成:中间体7(500mg,0.54mmol),9-蒽硼酸(360mg,1.62mmol,3.0equiv),pd(pph3)4(63mg,10mol%),na2co3(342mg,3.24mmol,6.0equiv),以及kbr
(193mg,1.62mmol,3.0equiv)置于100毫升双口烧瓶中,然后液氮冷却下往反应瓶中加入dme(6ml),h2o(2.0ml)。所得混合体系,于液氮冷却下氮气置换3次,以对溶剂进行充分脱气。脱气完毕后,将反应体系缓慢升温至室温,并在油浴中加热回流12h。反应结束后,冷却至室温,并加入30ml h2o,dcm萃取(3*20ml),有机相合并并用饱和实验水洗涤,无水硫酸钠干燥,过滤旋干得粗产物,最后经硅胶柱层析(石油醚:乙酸乙酯=30:1)得中间体8(淡黄色固体,410mg,77%yield)。
[0078]1h nmr(400mhz,cdcl3)δ8.52(s,2h),8.11
–
8.06(m,4h),8.01
–
7.99(m,4h),7.94(d,j=8.0hz,2h),7.78(d,j=8.0hz,2h),7.54
–
7.44(m,10h),7.40
–
7.33(m,8h),3.34(s,6h),3.10
–
3.01(m,4h),1.29
–
1.19(m,24h)ppm.
[0079]
13
c nmr(101mhz,cdcl3)δ155.0,147.6,147.4,138.3,138.1,134.9,134.1,133.9,131.6,131.0,130.5,130.34,130.32,128.5,128.1,127.3,127.0,126.4,126.2,126.04,125.98,125.9,125.34,125.27,125.2,125.1,124.9,60.0,31.2,31.1,25.60,25.55,23.4,23.3ppm.
[0080]
中间体9的合成:氮气保护下,将中间体8(410mg,0.42mmol)溶解于10ml超干的dcm中,并将所得溶液冷却至-78℃,并缓慢滴加bbr3(4.2ml,4.2mmol,1.0m in dcm,10equiv),滴加完毕后,将反应体系温度缓慢升至室温,并搅拌48h。反应结束后,往反应体系后缓慢加入20ml h2o,dcm萃取(3*20ml),将有机相合并后用饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋干得粗产物,最后经硅胶柱层析(石油醚:乙酸乙酯=50:1to 20:1)得中间体9(淡黄褐色固体,363mg,90%yield)。
[0081]1h nmr(400mhz,cdcl3)δ8.53(s,2h),8.11
–
8.05(m,4h),8.03
–
8.01(m,4h),7.90(d,j=8.0hz,2h),7.73(d,j=8.0hz,2h),7.54
–
7.43(m,12h),7.41
–
7.33(m,6h),5.17(s,2h),3.11
–
3.05(m,2h),2.95
–
2.88(m,2h),1.28(d,j=4.0hz,6h),1.22(d,j=4.0hz,6h),1.18(d,j=4.0hz,6h),1.13(d,j=4.0hz,6h)ppm.该化合物为已知化合物,氢谱数据与文献中报道的符合得很好。
[0082]
手性磷酸10的合成:氮气保护下,将中间体9(363mg,0.38mmol)溶解于超干的吡啶(10ml),将所得的溶液冷却至0℃,并缓慢滴加pocl3(583mg,3.8mmol,10equiv),滴加完毕后将反应体系在90℃油浴中加热24h。然后,将反应体系冷却至0℃,搅拌情况下,缓慢滴加水(5ml),滴加完毕后,再将反应体系升温至90℃并搅拌3h。反应完毕后,将反应体系冷却至室温,加入h2o(20ml),dcm萃取(3*20ml),有机相合并后先用4m hcl洗涤(3*20ml),饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋干得粗产物,最后硅胶柱层析(dcm:meoh=50:1)得手性磷酸催化剂10(浅黄褐色固体,349mg,90%yield)。
[0083]1h nmr(400mhz,cdcl3)δ8.45(s,2h),8.07
–
8.03(m,6h),7.88
–
7.81(m,4h),7.74
–
7.71(m,2h),7.59
–
7.55(m,2h),7.50
–
7.47(m,2h),7.43
–
7.39(m,4h),7.31
–
7.27(m,4h),7.17(s,2h),6.84
–
6.80(m,2h),6.60
–
6.56(m,2h),2.87
–
2.82(m,2h),2.76
–
2.72(m,2h),1.10
–
1.06(m,12h),0.94
–
0.92(m,6h),0.72
–
0.70(m,6h)ppm.
[0084]
31
p nmr(162mhz,cdcl3)δ3.65ppm.
[0085]
为了进一步解释与说明本发明的优势,以及本发明的方法与传统方法的区别,下面仅给出7在合成其他全新大位阻binol衍生物中的应用,但特别指出的是获得的中间体11和12都能容易地转化成相应的手性磷酸。
[0086]
具体应用实施例2:
[0087][0088]
按照合成中间体8相同的方法及投料比,全新骨架的大位阻手性磷酸中间体11可简单合成。
[0089]
白色固体(471mg,80%yield).
[0090]1h nmr(400mhz,cdcl3)δ7.91
–
7.89(m,2h),7.85
–
7.84(m,4h),7.80
–
7.79(m,4h),7.75
–
7.72(m,8h),7.56
–
7.54(m,4h),7.52
–
7.48(m,8h),7.47
–
7.32(m,10h),3.15(s,6h),3.00
–
2.87(m,4h),1.29(d,j=4hz,6h),1.25(d,j=4hz,6h),1.22(d,j=4hz,6h),1.15(d,j=4hz,6h)ppm.
[0091]
13
c nmr(101mhz,cdcl3)δ155.0,148.2,147.7,143.2,142.3,141.4,140.8,135.4,134.0,133.7,130.8,130.3,128.9,128.0,127.54,127.48,126.2,125.8,125.4,125.2,124.8,124.7,121.9,60.0,31.2,31.0,25.6,25.2,23.43,23.39ppm.
[0092]
hrms(es )calcd for c
82h75
o2[m h]
:1091.5767,found:1091.5751.
[0093]
具体应用实施例3:
[0094][0095]
按照合成中间体8相同的方法及投料比,全新骨架的大位阻手性磷酸中间体12可简单合成。
[0096]
黄褐色固体(569mg,73%yield).
[0097]1h nmr(400mhz,cdcl3)d 8.05-7.98(m,8h),7.92-7.82(m,6h),7.78-7.76(m,12h),7.52-7.33(m,30h),3.36(s,6h),3.12
–
3.03(m,4h),1.30
–
1.21(m,24h)ppm.
[0098]
13
c nmr(101mhz,cdcl3)δ155.0,147.6,147.4,141.9,140.9,140.3,138.5,138.3,
136.6,134.8,134.0,133.9,130.99,130.98,130.5,130.06,130.05,130.00,129.96,129.2,128.9,128.1,127.6,127.42,127.35,127.12,127.05,126.20,126.19,126.1,126.0,125.9,125.8,125.4,125.3,125.24,125.23,125.1,124.94,124.90,60.0,31.2,31.1,25.60,25.55,23.4,23.3ppm.
[0099]
hrms(maldi-tof)calcd for c
110h91
o2[m h]
:1443.7019,found:1443.7005.
[0100]
本发明提供了一种手性binol衍生的三氟甲磺酸酯化合物7的合成及应用。利用本发明所提供的方法,发明申请人进行了5次重复实验以评估在合成中间体8过程中本方法的重复性,五次实验收率均在75%以上,即反应具有非常好的重现性。此外,用传统的kumada偶联无法合成化合物11和12。
[0101]
以上所述实施例,并不用以限制本技术,凡在本技术的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。