1.本发明涉及有机合成和原料药的制备领域,具体涉及7-羟基-[1,2,4]三唑并[1,5-a]吡啶的制备新方法,该化合物可作为抗肿瘤药物妥卡替尼的关键中间体。
背景技术:
[0002]
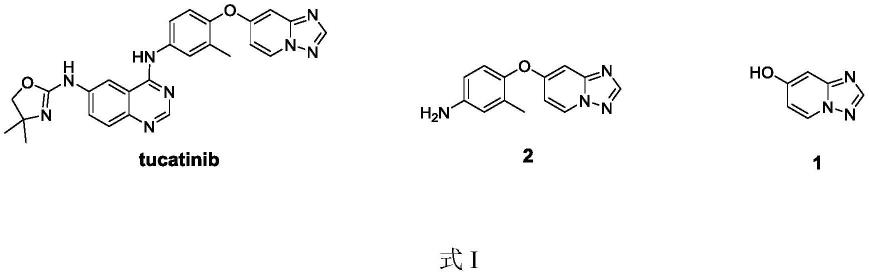
妥卡替尼,也称为irbinitinib,arry-380或ont-380,商品名为tukysa,是her2(人表皮生长因子受体酪氨酸激酶erbb-2)的口服抑制剂。由seattle genetics开发,并于2020年4月17日获得美国fda批准,用于治疗her2 乳腺癌。此外化合物1可以作为其他几类药物的中间体,包括her2和erbb的抑制剂(wo2021156180,wo2019241715,wo2019214651,wo2019214634)以及葡萄糖激酶(gka)激活剂(wo2013086397),
[0003]
7-羟基-[1,2,4]三唑并[1,5-a]吡啶(1)可以制备4-([1,2,4]三唑并[1,5-a]吡啶-7-基氧基)-3-甲基苯胺(2),为制备抗肿瘤药物妥卡替尼(tucatinib)的关键中间体,如式i所示:
[0004][0005]
目前关于化合物1的制备主要包括两种方法,如式ii所示:
[0006][0007]
方法一(wo 2008072634,wo 2019214651),以苄醇和2-硝基-4-氯吡啶(3)为原料,以thf为溶剂,在强碱nah的作用下,通过亲核取代得到化合物4-(苄氧基)-2-硝基吡啶(4),收率~60%;而后在pd催化下,以lihmds作碱用nh3取代cl得到化合物5;化合物5依次与dmf-dma、盐酸羟胺反应得到6,再经三氟乙酸酐或羟胺磺酸反应关环得到化合物7,通过pd/c催化氢解反应生成化合物1;后3步反应总收率~20%,产品需经柱层析纯化。
[0008]
方法二(wo 2019214634),以吡啶-2-甲酸为原料,经4步反应制备得到2-氨基-4-甲氧基吡啶(9),依次与dmf-dma、盐酸羟胺反应得到10,再经羟胺磺酸反应关环得到化合物11,通过吡啶盐酸盐高温熔融脱去甲氧基得到化合物1。起始原料9市场价格高,可以通过吡啶-2-甲酸经酯化、氯代、胺化、霍夫曼降解4步制备,总收率33%。
[0009]
上述两种合成方法的不足之处在于,路线步骤较长,且用到了多个类型的保护基,部分步骤需要柱层析纯化,不适合放大制备。因此,需要针对现有技术中所存在的缺陷,对现有技术加以改进,提供一种原料易得、工艺简洁、操作方便、收率更高的制备方法,以降低成本。
技术实现要素:
[0010]
本发明旨在提供一种制备7-羟基-[1,2,4]三唑并[1,5-a]吡啶的新方法。
[0011]
本发明技术方案为:
[0012]
一种有机中间体,7-羟基-[1,2,4]三唑并[1,5-a]吡啶,其化学结构如式1:
[0013][0014][0015]
上述有机中间体7-羟基-[1,2,4]三唑并[1,5-a]吡啶(1)的合成路线如式iii所示,制备方法包括以下步骤:
[0016][0017]
(1)n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺的制备:
[0018]
a.2-氨基-4-羟基吡啶与有机溶剂混合,加入n,n-二甲基甲酰胺二甲基缩醛(dmf-dma)至反应完全;
[0019]
b.加入盐酸羟胺至反应完全,得到n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺;
[0020]
(2)n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺与有机溶剂混合,加入三氟乙酸酐(tfaa),经分子内关环反应生成7-羟基-[1,2,4]三唑并[1,5-a]吡啶。
[0021]
步骤(1)a中,所述的有机溶剂为醇,尤其是c1-c4醇,优选为甲醇或者乙醇。
[0022]
步骤a反应条件为:70-95℃回流加热至反应完全。
[0023]
步骤b反应温度为20-95℃,优选为40-60℃。
[0024]
步骤(1)中,所述的2-氨基-4-羟基吡啶与n,n-二甲基甲酰胺二甲基缩醛的摩尔比为1:1-2,优选为1:1.1-1.9。盐酸羟胺与2-氨基-4-羟基吡啶的摩尔比为1:1.1-1.5,优选为1:1.2-1.3。
[0025]
步骤(2)中,所述的n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺与三氟乙酸酐的摩尔比为1:1-2,优选为1:1.1-1.3。
[0026]
步骤(2)包括:
[0027]
a.n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺与有机溶剂混合后,50-75℃下,加入三氟乙酸酐,反应0.5-10h;或者在5-15℃下搅拌反应0.5-1h后再回流反应20min-1h;
[0028]
b.去除溶剂,反应产物洗涤至中性;
[0029]
c.取固体洗涤干燥。
[0030]
步骤a中,所述的有机溶剂为四氢呋喃(thf)、乙腈、二甲亚砜(dmso)或者二甲基甲酰胺(dmf)。优选为四氢呋喃。
[0031]
优选的,步骤b中,去除溶剂后,用弱碱溶液洗涤反应产物至中性,所述的弱碱溶液为碳酸氢盐或碳酸盐溶液,更优选为饱和溶液。或者,步骤b中,去除溶剂后,用弱碱溶液及有机溶剂洗涤至中性,分液取有机相干燥。所述的有机溶剂为二氯甲烷,所述的弱碱溶液为碳酸氢盐或碳酸盐溶液,更优选为饱和溶液。
[0032]
步骤c中,用有机溶剂iii洗涤干燥,所述的有机溶剂iii为醇,优选为甲醇或乙醇。
[0033]
通过上述方法可以得到具有较高纯度的7-羟基-[1,2,4]三唑并[1,5-a]吡啶。
[0034]
步骤(1),2-氨基-4-羟基吡啶依次与n,n-二甲基甲酰胺二甲基乙缩醛和盐酸羟胺反应,不需要经过分离纯化即可得到n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺,产物纯度》99%;步骤(2)中,步骤(1)产物与三氟乙酸酐反应经分子内关环得到7-羟基-[1,2,4]三唑并[1,5-a]吡啶,在经过洗涤后,产物纯度》98%,后处理简单方便。各步骤的收率均超过80%,总收率可以达到70%。
[0035]
而且本发明的方法,只需要两部反应即可得到高纯度7-羟基-[1,2,4]三唑并[1,5-a]吡啶;步骤少,操作方便;并且步骤(1)不需要对产物进行纯化,工艺更为简洁;最终产品纯度高,后处理简单方便。
[0036]
与现有技术相比,本发明的方法原料易得,操作简单,反应条件温和;而且制备过程中,不需要引入保护基团,收率有显著提高,可以降低成本,适合工艺放大制备,可用于工业化大规模生产。
具体实施方式
[0037]
以下结合具体的实施例来对本发明的技术方案加以说明。
[0038]
实施例1 n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺(13)的制备(1)
[0039]
取250ml三口瓶,加入2-氨基-4-羟基吡啶(15g,0.136mol),再加入90ml etoh,搅拌,呈黄色悬浊液,加入dmf-dma(33g,0.245mol),80℃加热回流3h,tlc检测反应完全。关加热,待反应液冷却至40℃,投入12g盐酸羟胺(0.17mol),50℃下反应2h,悬浊液变为白色,tlc检测反应完全,停止加热,冷却至室温,抽滤,60℃烘干得类白色固体13(18.1g,收率87%)。
[0040]1h nmr(400mhz,dmso-d6)δ10.29(brs,1h),9.96(s,1h),9.08(d,j=2.6hz,1h),7.82(d,j=5.4hz,1h),7.78(d,j=9.6hz,1h),6.42(d,j=2.6hz,1h),6.95(dd,j=5.2hz,2.6hz,1h).
[0041]
13
c nmr(100mhz,dmso-d6)δ165.8,154.5,148.8,136.5,106.5,96.5.
[0042]
lc-ms(esi):154[m h]
.
[0043]
hplc:柱:agilent eclipse xdb-c18(250mm
×
4.6mm
×
5μm);检测:210nm;流速:0.8ml/min;温度:30℃;注射量:1μl;溶剂:甲醇;浓度:0.2mg/ml;运行时间:50min;流动相
a:水;流动相b:甲醇/三乙胺=100:0.1;洗脱梯度:流动相a/流动相b=10/90:tr=1.906min,纯度:99.69%。
[0044]
实施例2 n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺(13)的制备(2)
[0045]
将2-氨基-4-羟基吡啶(42.9g,0.39mol)加入到甲醇(150ml)中,在反应液中加入dmf-dma(51.0g,0.43mol),将反应悬浮液搅拌并加热至60-65℃反应2h,得到浅黄色溶液。将所得溶液冷却至约40℃,然后将盐酸羟胺(33.0g,0.47mol)添加至反应物中,将反应溶液搅拌并加热至50-55℃反应2h。将反应溶液冷却至室温,再用冰水浴冷却1h,生成固体,抽滤,用甲醇(15ml
×
1)洗涤,55℃下干燥8h,得到类白色固体13(49.5g,83%)。
[0046]
谱图检测结果同实施例1,纯度为99.10%。
[0047]
实施例3 7-羟基-[1,2,4]三唑并[1,5-a]吡啶(1)的制备(1)
[0048]
将n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺(13)(46.0g,0.3mol)加入到thf(380ml)中,搅拌并加热至60-65℃。在30min内将tfaa(75.6g,0.36mol)滴加到反应物中,控制在65-70℃。将反应物在65
–
70℃下再搅拌1h,得到浅黄色悬浮液。减压除去溶剂,得到黄色糊状物,向其中加入饱和碳酸氢钠溶液(600ml)至ph=7,并在5-10℃下搅拌1h。生成固体,抽滤,用水(20ml
×
2)洗涤,得到78g湿品。将其与甲醇(100ml)混合,搅拌并加热至回流30min,在5-10℃下搅拌1h。生成固体,抽滤,用甲醇(15ml
×
2)洗涤,在55℃下干燥6h,得到类白色固体1(36.7g,81%)。
[0049]1h nmr(400mhz,dmso-d6)δ8.79(d,j=5.2hz,1h),8.46(s,1h),7.06(d,j=2.6hz,1h),6.84(dd,j=5.2hz,2.6hz,1h).
[0050]
13
c nmr(100mhz,dmso-d6)δ162.8,150.0,148.6,131.2,110.1,95.5.
[0051]
lc-ms(esi):174.1[m na]
.
[0052]
hplc:柱:agilent eclipse xdb-c18(250mm
×
4.6mm
×
5μm);检测:210nm;流速:0.8ml/min;温度:30℃;注射量:1μl;溶剂:甲醇;浓度:0.2mg/ml;运行时间:40min;流动相a:水;流动相b:甲醇/三乙胺=100:0.1;洗脱梯度:流动相a/流动相b=10/90:tr=3.388min,纯度:98.39%。
[0053]
实施例4 7-羟基-[1,2,4]三唑并[1,5-a]吡啶(1)的制备(2)
[0054]
将n-羟基-n'-(4-羟基吡啶-2-基)-甲酰胺(13)(4.6g,0.03mol)加入到thf(50ml)中,冷至5℃。在10min内将tfaa(7.6g,0.036mol)滴加到反应物中,控制在5-10℃。将反应物在10
–
15℃下再搅拌1h,得到浅黄色悬浮液,加热至回流30min。减压除去溶剂,得到黄色糊状物,向其中加入二氯甲烷50ml、饱和碳酸氢钠溶液(50ml),搅拌分液,用水(50ml
×
2)洗涤有机相,有机相浓干得到7.1g粗品。将其与甲醇(20ml)混合,搅拌并加热至回流30min,在5-10℃下搅拌1h。生成固体,抽滤,用甲醇(5ml
×
2)洗涤,在55℃下干燥6h,得到浅黄色固体1(2.4g,53%)。
[0055]
谱图检测结果同实施例3,纯度为97.81%。
[0056]
需要指出的是,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项目技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。