1.本发明涉及杂环化合物的合成技术领域,尤其是涉及一种2,4-二氨基嘧啶氧化物的制备方法。
背景技术:
2.2,4-二氨基嘧啶氧化物,商业名称为亚美尼斯分子(aminexil),是欧莱雅集团研发的独家专利防脱成分,广泛用于其洗护发产品中。许多脱发案例往往伴随有“毛囊周纤维化”的情况,即毛囊周边的胶原纤维变厚、变硬,使得毛球向真皮的深层插入受阻,致使毛囊受到挤压,无法输送营养物质,导致发丝越来越脆弱、细软。2,4-二氨基嘧啶是一种相对安全的固发成分,能够有效软化毛囊周边的角质,延长毛囊寿命;而且无副作用,坚持使用可达到长效防脱的目的。
3.us3644364、us3382247、cn87104693等报道了嘧啶衍生物的制备方法,但均直接以二氨基卤代嘧啶作为底物,产物均为米诺地尔(minoxidil)的衍生物,并未提出亚美尼斯分子的合成途径。
4.us2416617、wo2008/98058、wo2007/84786、wo2012/109423报道合成制备2,4-二氨基嘧啶(亚美尼斯分子前体),主要采用卤代氮杂环前体经过氨取代合成,原料复杂昂贵,合成反应条件苛刻。早期文献报道采用3,3-二乙氧基丙腈与盐酸胍可以缩合合成2,4-二氨基嘧啶(journal of the american chemical society,1950,72,2587-2593)。但3,3-二乙氧基丙腈的制备需要用乙腈、甲醇钠、甲酸乙酯为原料,在40-50个大气压的高压条件下与一氧化碳反应(us4525310),该方法反应条件苛刻,危险性和设备要求高。
5.有鉴于此,特提出本发明。
技术实现要素:
6.本发明的目的在于提供一种2,4-二氨基嘧啶氧化物的制备方法。
7.为了实现本发明的上述目的,特采用以下技术方案:本发明提供了一种2,4-二氨基嘧啶氧化物的制备方法,所述方法包括以下步骤:(1)3-羟基丙腈经氧化获得3-氧代丙腈;(2)3-氧代丙腈和胍在氧化条件下进行环合反应,得到2,4-二氨基嘧啶氧化物。
8.本发明设计出一种全新的亚美尼斯分子合成路径,由3-氧代丙腈和胍(如盐酸胍)在氧化条件下生成亚美尼斯分子。其中,3-氧代丙腈由廉价易得的医药化学品3-羟基丙腈为原料,经氧化后得到。
9.合成路线为:
10.步骤(1)氧化过程可以采用化学氧化剂或生物氧化酶作为氧化剂来进行,即可以在化学氧化剂存在下进行氧化,也可以在生物氧化酶存在下进行氧化,没有特别限制,可以根据不同的氧化剂选择适合的条件制备得到3-氧代丙腈(3-oxo-propionitrile,也可称为丙腈醛)。
11.在一些实施方式中,步骤(1)包括:将3-羟基丙腈在化学氧化剂存在下进行氧化获得3-氧代丙腈。
12.化学氧化剂可以为本领域已知的用于将醇氧化成醛的化学试剂,包括但不限于双氧水、kmno4、kclo3、浓h2so4、hno3、mno2、fecl3等,优选为双氧水。
13.化学氧化剂与3-羟基丙腈的摩尔比可以为5:1-15:1(例如5:1、6:1、8:1、10:1、12:1、15:1);在化学氧化剂存在下进行氧化的过程可以在加热环境中进行。加热温度可以为40-60 ℃(例如40、50、60℃),反应时间可以为2-6 h(例如2、3、4、5、6 h)。
14.在一些实施方式中,步骤(1)包括:将3-羟基丙腈在生物氧化酶存在下进行氧化获得3-氧代丙腈。
15.生物氧化酶可以为本领域已知的具有醛耐受性的广谱短链醇(一般指c1-c6醇)脱氢酶和氧化酶,例如乙醇脱氢酶、乙醇氧化酶。
16.在生物氧化酶存在下进行氧化的过程可以在溶液中进行。溶液的ph可以控制在7-8,优选为约7.5。这可以通过,例如采用ph约7.5的磷酸盐缓冲剂而实现。
17.在一些实施方式中,上述步骤(1)包括将获得的3-氧代丙腈进行提纯的步骤。将3-氧代丙腈提纯进行下一步反应可以提高终产物纯度。
18.提纯可以包括如下步骤:3-氧代丙腈用正丁醇溶解,离心,上清用无水硫酸钠干燥,加入浓硫酸(调节ph)和任选的无水硫酸铜(促进成环),密封,加热过夜,取出离心留上清液,用碳酸氢钠水溶液萃取,上层有机相再用无水硫酸钠干燥。
19.步骤(2)本步骤中,3-氧代丙腈和胍在氧化条件下发生环合反应得到2,4-二氨基嘧啶氧化物。
20.为了更微观地反映反应过程,该步骤可以拆解为两个步骤,反应前后n的变化如下:
21.胍可以采用胍的盐酸盐(盐酸胍)、胍的硝酸盐(硝酸胍)等,优选盐酸胍。
22.此时可以在反应体系中加入碱以将盐酸胍转化为胍,并促进环合反应。
23.碱可以为本领域已知的用作碱催化的试剂,包括但不限于碳酸锂、氢氧化锂、叔丁醇锂、碳酸钠、碳酸氢钠、氢氧化钠、磷酸钠、甲醇钠、乙醇钠、异丙醇钠、叔丁醇钠、碳酸钾、碳酸氢钾、氢氧化钾、磷酸钾、甲醇钾、乙醇钾、叔丁醇钾、碳酸铯、氢氧化铯、碳酸镁、氢氧化镁、磷酸镁、氧化镁、甲醇镁、乙醇镁、异丙醇镁、叔丁醇镁、三乙胺、二异丙基胺、二异丙基乙基胺、三正丁胺、吡啶、2-甲基吡啶、2,6-二甲基吡啶、4-二甲氨基吡啶、四氢吡咯、吗啉、哌啶或2,2,6,6-四甲基哌啶中的一种或其组合物,优选为碳酸钠、氢氧化钠、碳酸钾、碳酸氢钾或氢氧化钾等。
24.环合反应可以在溶剂中进行。
25.溶剂可以为本领域已知的能够作为3-氧代丙腈和胍反应介质且不与3-氧代丙腈和胍发生反应的溶剂,包括但不限于甲醇、乙醇、异丙醇、正丁醇、异丁醇、异戊醇、二氯甲烷、氯仿、苯、甲苯、二甲苯、氯苯、乙腈、四氢呋喃、2-甲基四氢呋喃、二氧六环、乙二醇二甲醚、甲基叔丁基醚、醋酸异丙酯、醋酸正丁酯、苯基甲基醚、乙醇、异丙醇等。
26.氧化条件可以在空气中自发氧化,以及制备3-氧代丙腈的过程中有残留的少量氧化剂,也会加速氧化的过程,在缩合的同时氧化就会发生。
27.在一些实施方式中,胍和3-氧代丙腈的摩尔比为1:1-5:1(例如1:1、2:1、3:1、4:1、5:1),优选为3:1。
28.若步骤(1)进行了提纯,这里的3-氧代丙腈指提纯后的3-氧代丙腈。
29.通过控制两者的摩尔比,可以使反应物转化最完全,如果小于或大于该比例,会有大量原料剩余,导致经济性降低。
30.在一些实施方式中,步骤(2)中反应温度为70-85℃,优选为77℃;反应时间为20-30min。将两个底物(盐酸胍和3-氧代丙腈)混合后,最快在20分钟就可以看到产物,在30分钟时反应基本完全。
31.反应温度控制对反应速率有影响,如果小于或大于该反应温度,反应时间将有所延长,或产物含量降低。
32.在一些实施方式中,二氨基嘧啶氧化物的制备方法包括以下步骤:取3-羟基丙腈置于离心管中,加入化学氧化剂,化学氧化剂与3-羟基丙腈的摩尔比为5:1-15:1,50℃加热4 h,分装后冻干6 h,-20℃密封保存备用,得到3-氧代丙腈冻干物;取3-氧代丙腈冻干物用正丁醇溶解,离心,上清用无水硫酸钠干燥,加入浓硫酸,密封,加热过夜,取出离心留上清液,用等体积碳酸氢钠水溶液萃取,上层有机相再用无水硫酸钠干燥,得到提纯后的3-氧代丙腈;随后加入盐酸胍和乙醇钠乙醇溶液,盐酸胍和提纯
后的3-氧代丙腈的摩尔比为1:1-5:1,密封,77℃加热30 min。
33.在又一些实施方式中,二氨基嘧啶氧化物的制备方法包括以下步骤:将含3-羟基丙腈的底物溶液与生物氧化酶混合,摇床反应,干燥,获得3-氧代丙腈;取3-氧代丙腈冻干物用正丁醇溶解,离心,上清用无水硫酸钠干燥,加入无水硫酸铜和浓硫酸,密封,加热过夜,取出离心留上清液,无水硫酸钠干燥,得到提纯后的3-氧代丙腈;随后加入盐酸胍和乙醇钠乙醇溶液,盐酸胍和提纯后的3-氧代丙腈的摩尔比为1:1-5:1,密封,77℃加热30 min。
34.有益效果本发明的反应绿色温和,实现从简单、价廉、易得的原料出发合成亚美尼斯分子。
35.在上文中已经详细地描述了本发明,但是上述实施方式本质上仅是例示性,且并不欲限制本发明。此外,本文并不受前述现有技术或发明内容或以下实施例中所描述的任何理论的限制。
36.除非另有明确说明,在整个申请文件中的数值范围包括其中的任何子范围和以其中给定值的最小子单位递增的任何数值。除非另有明确说明,在整个申请文件中的数值表示对包括与给定值的微小偏差以及具有大约所提及的值以及具有所提及的精确值的实施方案的范围的近似度量或限制。除了在详细描述最后提供的工作实施例之外,本技术文件(包括所附权利要求)中的参数(例如,数量或条件)的所有数值在所有情况下都应被理解为被术语“大约”修饰,不管“大约”是否实际出现在该数值之前。“大约”表示所述的数值允许稍微不精确(在该值上有一些接近精确;大约或合理地接近该值;近似)。如果“大约”提供的不精确性在本领域中没有以这个普通含义来理解,则本文所用的“大约”至少表示可以通过测量和使用这些参数的普通方法产生的变化。例如,“大约”可以包括小于或等于10%,小于或等于5%,小于或等于4%,小于或等于3%,小于或等于2%,小于或等于1%或者小于或等于0.5%的变化。
附图说明
37.图1示出本发明实施例1制得的3-氧代丙腈的色谱图。
38.图2示出本发明实施例1制得的3-氧代丙腈的质谱图。
39.图3示出本发明实施例3制得的二氨基嘧啶氧化物的色谱图。
40.图4示出本发明实施例3制得的二氨基嘧啶氧化物的质谱图。
41.图5示出本发明实施例4制得的二氨基嘧啶氧化物的色谱图。
42.图6示出本发明实施例4制得的二氨基嘧啶氧化物的质谱图。
具体实施方式
43.下面结合实施例对本发明作进一步的说明,需要说明的是,提供以下实施例仅出于说明目的并不构成对本发明要求保护范围的限制。
44.除特殊说明外,在实施例中所采用的原料、试剂、方法等均为本领域常规的原料、试剂、方法。
45.色谱仪使用安捷伦1200。
46.质谱仪使用qtrap 4500三重四极杆质谱(sciex公司)。
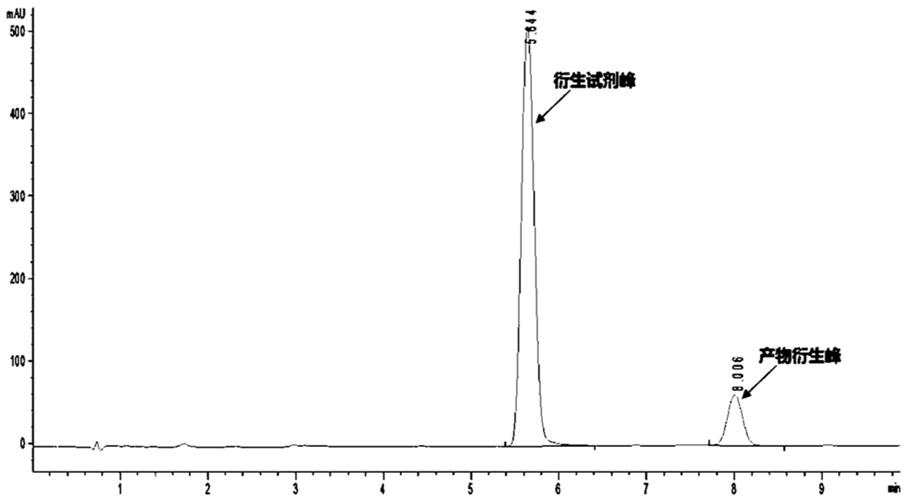
47.实施例1 3-氧代丙腈合成取200 μl 3-羟基丙腈置于50 ml离心管中,加入1.6 ml 30%双氧水,加入60 μl三氯化铁水溶液(800 mm),50℃加热4 h,分装3管后冻干6 h,-20℃密封保存备用。
48.样品检测:样品用300 μl正丁醇溶解,离心,取2 μl水稀释100倍,吸取10
ꢀµ
l稀释液加10
ꢀµ
l dnph衍生试剂于60℃衍生30分钟再加20
µ
l乙腈,进液相色谱和质谱检测。
49.dnph衍生试剂配制方法:4 mg dnph(2,4-二硝基苯肼)溶于4 ml溶液中(1 ml浓盐酸,0.5 ml乙腈,2.5 ml水)超声溶解,-20℃保存备用。
50.色谱条件:色谱柱:c18反向柱;流动相:a(0.1%的甲酸水):b(乙腈)=40:60;流速:1ml/min;检测波长:360nm;保留时间:8.0min。
51.检测结果:色谱图如图1所示。质谱图如图2所示。证明产物中含有3-氧代丙腈。
52.实施例2 3-氧代丙腈合成1、生物氧化酶制备:取毕赤酵母(pichia pastoris)菌株甘油管菌液在ypd固体培养基上进行划线活化,37℃培养3天。在ypd平板中挑一个单克隆,接到10ml ypd液体培养基中,30℃培养24小时左右,按2%-5%的接种量转接到250ml ypm液体培养基中,待od=1时,用甲醇(按培养基体积的1%)进行诱导,诱导48小时。菌生长至od
600nm
≈8-9,将菌液分装收到50ml离心管中,5000rpm离心10分钟,弃上清,菌体沉淀放-20℃备用。
53.培养基配置:ypd培养基的配制(1l):10g酵母粉;20g蛋白胨;20g 葡萄糖;溶解至于1l体积,115℃灭菌30min。固体培养基中需添加1.5%的琼脂。
54.ypm培养基的配制(1l):10g酵母粉,20g蛋白胨,50g麦芽糖。115℃灭菌30min。
55.用2-3ml 0.25m ph7.5磷酸缓冲液重悬酵母菌体,菌浓度为od
600nm
≈75,在超声破碎仪中破碎细胞(功率30%,破碎时间10min),然后在8000rpm/min离心10min,弃菌体碎片沉淀,将上清分装到2ml离心管中,分别加入1.5倍体积的丙酮,冷藏30min,12000rpm/min离心2min,弃上清,蛋白沉淀中加入相当于初始细胞破碎清液1/2体积的0.25m的磷酸缓冲液进行复溶,12000rpm/min离心2min,弃去不溶物,上清中含有以醇为底物的氧化酶(氧化酶溶液),分装后-20℃冰箱保存备用。
56.2、合成3-氧代丙腈以3-羟基丙腈为底物,采用上述步骤1制备获得的含氧化酶的上清液,对底物羟基进行氧化成醛。
57.表1
按表1中给出比例配置成10ml底物溶液,底物溶液与上述氧化酶溶液按1:1体积比混合后于30℃摇床反应4小时,取样检测产物3-氧代丙腈含量,检测方法同实施例1,生成2.1mm产物3-氧代丙腈,冻干6 h,-20℃密封保存备用。
58.实施例3 二氨基嘧啶氧化物的合成取一管3-氧代丙腈冻干物(实施例1制备),样品用300 μl正丁醇溶解,离心,上清用无水硫酸钠干燥3次,加入9
ꢀµ
l浓硫酸(调节ph),密封,77℃加热过夜,取出离心留上清液,用等体积0.2 m碳酸氢钠水溶液萃取3次,上层有机相再用无水硫酸钠干燥3次,得到提纯后的3-氧代丙腈,随后加入10 mg盐酸胍(盐酸胍和提纯后的3-氧代丙腈的摩尔比为3:1)和50
ꢀµ
l 20%的乙醇钠乙醇溶液,密封,77℃加热30 min,检测产物。
59.样品检测:样品用水稀释50倍直接进lc-ms检测。
60.色谱条件:色谱柱:c18反向柱;流动相:a(0.1%的甲酸水):b(乙腈)=98:2;流速:1 ml/min;检测波长:254 nm;保留时间:3.7 min。
61.检测结果:如图3和图4所示,产物大部分为氧化物,也有一小部分未氧化。
62.实施例4 二氨基嘧啶氧化物的合成取一管3-氧代丙腈冻干物(实施例2制备),样品用300 μl正丁醇溶解,离心,上清用无水硫酸钠干燥3次,加入30 mg无水硫酸铜(促进成环反应)和9
ꢀµ
l浓硫酸(调节ph),密封,77℃加热过夜,取出离心留上清液,无水硫酸钠干燥2次,加入10 mg盐酸胍和80
ꢀµ
l 20%的乙醇钠乙醇溶液,密封,77℃加热30 min,检测产物。
63.样品检测:样品用水稀释50倍直接进lc-ms检测。
64.检测条件:同实施例3。
65.检测结果:如图5和图6所示,产物大部分为氧化物,也有一小部分未氧化。
66.以上各实施例仅用以举例说明本发明的技术方案,而非对其限制。尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:在没有脱离本发明权利要求所限定的精神和实质的范围内,可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换仍然在本发明权利要求所限定的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。