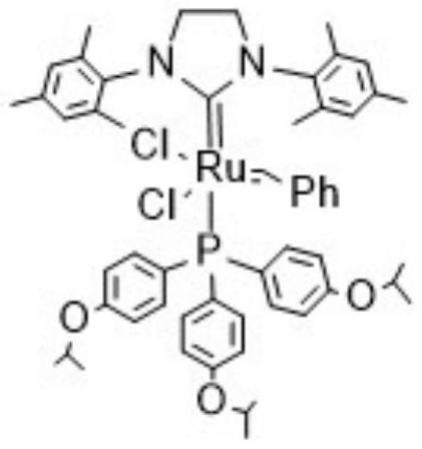
1.本发明涉及钌卡宾结构化合物及其合成技术领域,具体地说,涉及一种钌卡宾催化剂及其制备方法,以及在催化开环易位复分解反应中的应用。
背景技术:
2.烯烃复分解反应是两个断裂的烯烃碳碳双键重新组合成新碳碳双键的过程。在天然产物的合成、材料科学中均有广泛的应用。
3.对烯烃复分解反应影响最大的因素是催化剂。多年来,为寻找高效、专一的催化剂一直是烯烃复分解的研究焦点。鉴于schrock、grubbs、chauvin三人在烯烃复分解催化剂上做出的贡献,共同获得2005年度诺贝尔化学奖。
4.在烯烃复分解催化剂体系中,应用最广且最实用的就是grubbs催化剂(格拉布催化剂),一种钌卡宾结构的化合物,而因其对氧和质子性溶剂的稳定性在制药及材料行业具有越来越广泛的应用。又由于其对含质子的极性基团很好的耐受性,在各种需要利用烯烃复分解反应构建新的大环结构分子的合成上有着重要的作用。
技术实现要素:
5.本发明旨在提供一种钌卡宾催化剂及其制备方法,以及在催化开环易位复分解反应中的应用。本发明的卡宾结构钌催化剂由钌卡宾催化剂与吡啶反应获得含吡啶的钌卡宾催化剂后,1-溴-4-异丙氧基苯与镁和三氯化磷反应获得三(4-异丙氧基苯)膦后,含吡啶的钌卡宾催化剂与三(4-异丙氧基苯)膦反应即可形成所需的钌卡宾催化剂。
6.本发明的技术方案具体如下:
7.本发明的第一目的在于提供了一种钌卡宾催化剂,具有如下所示的结构式:
[0008][0009]
本发明的第二目的在于提供上述钌卡宾催化剂的制备方法,包括:
[0010]
(1)将grubbs二代催化剂(ⅱ)和吡啶类化合物于有机溶剂中溶解混合,经搅拌反应后,经后处理得到绿色的含吡啶的钌卡宾催化剂(ⅲ);
[0011]
其中,所述grubbsii催化剂结构如式(ⅱ)所示:
[0012][0013]
所述含吡啶的钌卡宾催化剂结构如式(ⅲ)所示:
[0014]
其中r=h、ch3[0015][0016]
(2)将1-溴-4-异丙氧基苯和碎镁屑置于有机溶剂中,于室温下回流反应2小时后,向反应液中加入三氯化磷继续加热回流2小时后,萃取反应液即可得到配体三(4-异丙氧基苯)膦(ⅳ);
[0017]
其中,所述配体三(4-异丙氧基苯)膦结构如式(ⅳ)所示:
[0018][0019]
(3)将步骤(1)制备的含吡啶的钌卡宾催化剂(ⅲ)用有机溶剂溶解后加入步骤(2)制备的配体三(4-异丙氧基苯)膦(ⅳ),然后搅拌反应至溶液颜色呈棕色,经后处理制得如式(ⅰ)所示的钌卡宾结构化合物。
[0020]
进一步的,所述步骤(1)中的grubbs二代催化剂(ⅱ)与吡啶类化合物的质量比为1:1-5;优选的质量比为1:1-2。
[0021]
进一步的,所述步骤(1)中的搅拌反应温度为10-40℃,反应时间10min-1h;优选反应温度5-25℃,反应时间10-20min。
[0022]
进一步的,所述步骤(1)中的有机溶剂选自二氯甲烷、三氯甲烷、四氯甲烷、四氢呋喃、三氯苯、甲苯或苯中的一种或几种的混合物;优选的有机溶剂为甲苯或苯。
[0023]
进一步的,所述步骤(1)中的后处理,具体操作为:将搅拌后的混合物通过套管转移至低温溶剂中,依次经沉淀、过滤和洗涤,真空下干燥后得含吡啶的钌卡宾催化剂(ⅲ);其中,所述溶剂为碳原子数小于20的烷烃类、环烷烃类或芳烃类化合物;优选溶剂为戊烷或己烷。
[0024]
进一步的,所述步骤(2)中的1-溴-4-异丙氧基苯、镁和三氯化磷的摩尔比为1-10:1-10:1;优选的摩尔比为3-4:3-4:1。
[0025]
进一步的,所述步骤(2)中有机溶剂选自无水乙醚、四氢呋喃、二氧六环、二甲亚砜中的一种或几种的混合物;优选的溶剂为无水乙醚。
[0026]
进一步的,所述步骤(2)中,将1-溴-4-异丙氧基苯和碎镁屑置于有机溶剂中,加热回流反应2小时后缓慢降至室温,于-100-0℃的环境温度下向反应液中缓慢滴加三氯化磷
后,再继续加热回流2小时,萃取反应液即可得到配体三(4-异丙氧基苯)膦(ⅳ)。
[0027]
进一步的,所述步骤(2)中的萃取所用萃取剂为碳原子数小于20的烷烃类、环烷烃类或芳烃类化合物;优选的萃取剂为无水乙醚。
[0028]
进一步的,所述步骤(3)中的含吡啶的钌卡宾催化剂(ⅲ)与配体三(4-异丙氧基苯)膦(ⅳ)的摩尔比为1:1-5;优选的摩尔比为1:1-1.5。
[0029]
进一步的,所述步骤(3)中的有机溶剂选自二氯甲烷、三氯甲烷、四氯甲烷、四氢呋喃、三氯苯、甲苯或苯中的一种或几种的混合物;优选溶剂为甲苯或苯。
[0030]
进一步的,所述步骤(3)中的后处理,具体操作为:将搅拌后的溶液真空除去溶剂,将所得残余物用溶剂洗涤,再经真空干燥后得式(ⅰ)所示的钌卡宾结构化合物;其中,所述溶剂为碳原子数小于20的烷烃类、环烷烃类或芳烃类化合物;优选溶剂为戊烷或己烷。
[0031]
进一步的,所述步骤(3)中的搅拌反应温度为5-30℃,反应时间为5min-1h;优选反应温度15-25℃,反应时间5-20min。
[0032]
进一步的,所述制备方法,所有步骤均在氮气保护下进行。
[0033]
本发明的第三目的在于提供上述钌卡宾催化剂在催化烯烃开环易位反应、闭环易位反应或交叉复分解等反应中的应用。
[0034]
进一步的,所述应用方法中的烯烃开环易位,具体包括:将上述钌卡宾催化剂溶于有机溶剂得混合液a,将双环戊二烯、改性剂及抗氧剂溶于有机溶剂得混合液b,将混合液a和混合液b充分混匀后,立即注入模具,于40℃~80℃发生烯烃开环易位反应,固化成型后,120℃~150℃进行后处理。
[0035]
进一步的,所述应用方法中的有机溶剂选自二氯甲烷、甲苯、乙酸乙酯或四氢呋喃中的一种或几种的混合物。
[0036]
进一步的,所述双环戊二烯与钌卡宾催化剂的物质的量比例为5000-50000:1。
[0037]
进一步的,所述烯烃开环易位反应的温度为50-70℃,反应时间为5-10分钟。
[0038]
以下为本发明的反应机理:
[0039][0040]
其中,r=h或ch3。
[0041]
与现有技术相比,本发明的有益效果为:
[0042]
本发明合成的新型卡宾结构钌催化剂带有三(4-烷氧苯基)膦配体,在催化聚合的过程中,相比以前的钌金属卡宾结构催化剂,三(4-烷氧苯基)膦配体更容易脱落,形成催化剂活性中心,提高了催化剂的引发速率,加快了反应速度。
具体实施方式
[0043]
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干调整和改进。这些都属于本发明的保护范围。
[0044]
以下实施例中所用原料均可通过市售获得。
[0045]
实施例1
[0046]
1#钌卡宾催化剂及其合成方法:
[0047]
在氮气保护下,将二代grubbs催化剂(购自安耐吉化学有限公司)(4.0g)溶解在甲苯(10ml)中,并加入吡啶(30ml)。于室温下搅拌反应10分钟,在此期间观察到颜色从红色变为亮绿色。将反应混合物通过套管转移至100ml冷(-10℃)戊烷中,并沉淀出绿色固体。过滤沉淀物,用4
×
50ml戊烷洗涤,并在真空下干燥,得到绿色粉末含吡啶的中间产物。
[0048]
氮气保护下,在干燥过的250m l三口瓶上迅速装好恒压滴液漏斗和回流冷凝管,向三口瓶中加入4.8g碎镁屑并加入50ml无水乙醚覆盖住,滴加3.8ml 1-溴-4-异丙氧基苯,加一粒碘,加热回流。当碘的颜色褪去变为灰色混浊液后,将36.2g 1-溴-4-异丙氧基苯和100ml无水乙醚配成混合溶液,置于恒压滴液漏斗中,慢慢滴入少量混合溶液于反应瓶中,放热回流,滴加完毕后,加热回流2小时。将上述反应瓶置于-78℃,缓慢滴加7.3g三氯化磷和20ml无水乙醚的混合溶液,滴加完毕后加热回流2小时,慢慢降至室温,用氯化铵水溶液水解,用分流漏斗分出有机相,水层用乙醚萃取,合并有机相和萃取液,无水硫酸镁干燥,减压蒸去乙醚,用油泵减压蒸馏,收集馏分160-200℃,得到磷配体。
[0049]
氮气保护下,将中间产物(150mg)和膦配体(90mg)在苯(10ml)中合并于室温下搅拌10分钟。真空除去溶剂,并将所得的棕色残余物用4
×
20ml的戊烷洗涤并真空干燥。获得为褐色粉末的复合物。
[0050]
该产物的谱图分析数据:
[0051]
分子式:c
55h65
n2o3pcl2ru
[0052]
m/z:1004.67(100%)
[0053]
elemental analysis:c:66.09;h:6.39;p:3.02
[0054]
hnmr(400mhz):
[0055]
δ=7.52-7.44(5h,m);4.15-4.76(39h,m);2.23-2.75(21h,m)。
[0056]
实施例2
[0057]
2#钌卡宾催化剂及其合成方法:
[0058]
在氮气保护下,将二代grubbs催化剂(4.0g)溶解在甲苯(10ml)中,并加入2-甲基吡啶(32ml)。于室温下搅拌反应15分钟,在此期间观察到颜色从红色变为亮绿色。将反应混合物通过套管转移至100ml冷(-10℃)戊烷中,并沉淀出绿色固体。过滤沉淀物,用4
×
50ml戊烷洗涤,并在真空下干燥,得到绿色粉末含吡啶的中间产物。
[0059]
氮气保护下,在干燥过的250m l三口瓶上迅速装好恒压滴液漏斗和回流冷凝管,向三口瓶中加入5.0g碎镁屑并加入50ml无水乙醚覆盖住,滴加3.8ml 1-溴-4-异丙氧基苯,加一粒碘,加热回流。当碘的颜色褪去变为灰色混浊液后,将36.2g 1-溴-4-异丙氧基苯和100ml无水乙醚配成混合溶液,置于恒压滴液漏斗中,慢慢滴入少量混合溶液于反应瓶中,放热回流,滴加完毕后,加热回流2小时。将上述反应瓶置于-78℃,缓慢滴加8.1g三氯化磷和20ml无水乙醚的混合溶液,滴加完毕后加热回流2小时,慢慢降至室温,用氯化铵水溶液水解,用分流漏斗分出有机相,水层用乙醚萃取,合并有机相和萃取液,无水硫酸镁干燥,减压蒸去乙醚,用油泵减压蒸馏,收集馏分160-200℃,得到磷配体。
[0060]
氮气保护下,将中间产物(163mg)和膦配体(98mg)在苯(10ml)中合并于室温下搅拌5分钟。真空除去溶剂,并将所得的棕色残余物用4
×
20ml的戊烷洗涤并真空干燥。获得为褐色粉末的复合物。
[0061]
该产物的谱图分析数据:
[0062]
分子式:c
55h65
n2o3pcl2ru
[0063]
m/z:1004.67(100%)
[0064]
elemental analysis:c:65.78;h:6.60;p:3.13
[0065]
hnmr(400mhz):
[0066]
δ=7.52-7.44(5h,m);4.15-4.76(39h,m);2.23-2.75(21h,m)。
[0067]
实施例3
[0068]
3#钌卡宾催化剂及其合成方法:
[0069]
在氮气保护下,将二代grubbs催化剂(4.0g)溶解在甲苯(10ml)中,并加入2,6-二甲基吡啶(34ml)。于室温下搅拌反应15分钟,在此期间观察到颜色从红色变为亮绿色。将反应混合物通过套管转移至100ml冷(-10℃)戊烷中,并沉淀出绿色固体。过滤沉淀物,用4
×
50ml戊烷洗涤,并在真空下干燥,得到绿色粉末含吡啶的中间产物。
[0070]
氮气保护下,在干燥过的250m l三口瓶上迅速装好恒压滴液漏斗和回流冷凝管,向三口瓶中加入5.3g碎镁屑并加入50ml无水乙醚覆盖住,滴加3.8ml 1-溴-4-异丙氧基苯,加一粒碘,加热回流。当碘的颜色褪去变为灰色混浊液后,将36.2g 1-溴-4-异丙氧基苯和100ml无水乙醚配成混合溶液,置于恒压滴液漏斗中,慢慢滴入少量混合溶液于反应瓶中,放热回流,滴加完毕后,加热回流2小时。将上述反应瓶置于-78℃,缓慢滴加7.3g三氯化磷和20ml无水乙醚的混合溶液,滴加完毕后加热回流2小时,慢慢降至室温,用氯化铵水溶液水解,用分流漏斗分出有机相,水层用乙醚萃取,合并有机相和萃取液,无水硫酸镁干燥,减压蒸去乙醚,用油泵减压蒸馏,收集馏分160-200℃,得到磷配体。
[0071]
氮气保护下,将中间产物(167mg)和膦配体(102mg)在苯(10ml)中合并于室温下搅拌20分钟。真空除去溶剂,并将所得的棕色残余物用4
×
20ml的戊烷洗涤并真空干燥。获得为褐色粉末的复合物。
[0072]
该产物的谱图分析数据:
[0073]
分子式:c
55h65
n2o3pcl2ru
[0074]
m/z:1004.67(100%)
[0075]
elemental analysis:c:65.86;h:6.50;p:3.14
[0076]
hnmr(400mhz):
[0077]
δ=7.52-7.44(5h,m);4.15-4.76(39h,m);2.23-2.75(21h,m)。
[0078]
应用实施例1-6
[0079]
1#钌卡宾催化剂在催化制备双环戊二烯中的应用
[0080]
以下应用实施例中的双环戊二烯与1#钌卡宾催化剂物质的量比例分别为5000:1、10000:1、15000:1、20000:1、30000:1、50000:1时双环戊二烯均聚物的制备方法。制备所得双环戊二烯均聚物的拉伸(gb/t2567-2008),弯曲性能(gb/t2567-2008),冲击强度(gb/t2567-2008),断裂伸长率(gb/t2567-2008)以及热变形温度性能(iso 75-1987)如表1所示。
[0081]
下面以双环戊二烯与1#钌卡宾催化剂,两者物质的量比例为5000:1(质量比约为800:1)为例,进行具体催化反应说明:
[0082]
(1)配置催化剂溶液a
[0083]
在手套箱中称取实施例1制备的钌卡宾催化剂(424.5mg,0.5mmol),将其溶解在10.6ml二氯甲烷中,制备成40mg/ml的催化剂溶液。
[0084]
(2)配置双环戊二烯溶液b
[0085]
双环戊二烯(纯度大于95%)事先加入过量的氢化钙,在80℃下氮气保护下搅拌12h,减压蒸馏进行精制,然后加入双环戊二烯质量2%-5%的二氯甲烷溶剂提前充分溶解。在氮气保护下在双环戊二烯溶液中加入双环戊二烯质量0.01%-2%抗氧剂(邻二叔丁基对甲基苯酚),于低温反应浴上充分搅拌均匀,控制温度在10-30℃。
[0086]
(3)混合固化成型
[0087]
量取3.85mla溶液,120gb溶液,将a液与b液在氮气保护下快速混合,待混合均匀后。快速注入反应模具中,模具开始进行程序升温,保持反应温度在60℃,恒温1h,后处理温度为140℃,保温1h。
[0088]
待模具冷却后,产品脱模,即得到两个模腔,分别为60g的产品。
[0089]
表1单体与催化剂不同比例下的聚合物力学性能
[0090][0091]
以上已对本发明创造的较佳实施例进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明创造精神的前提下还可作出种种的等同的变型或替换,这些等同的变型或替换均包含在本技术权利要求所限定的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。