1.本发明属于生物信息技术领域,具体涉及一种基于宏基因组学的毒素基因丰度检测方法,还涉及一种毒素基因的注释数据库构建方法。
背景技术:
2.毒素因子(virulence factor,vfs)指由细菌、病毒和真菌代谢产生的带有侵袭力和毒素等毒力性质的分子。微生物感染宿主,主要是因为相关的致病菌携带了可引起宿主细胞损伤的毒素因子编码基因,可抑制或逃避宿主的免疫反应,进而能够出入宿主细胞,并进一步和宿主掠夺营养,达到自身增殖生长的目的。
3.前沿科学研究中,常见的毒素因子数据库包括tadb(https://bioinfo-mml.sjtu.edu.cn/tadb/)、tox-prot(http://www.expasy.org/sprot/tox-prot)、t3db(http://www.t3db.ca/)和vfdb(http://www.mgc.ac.cn/vfs/)等。其中,vfdb数据库的总体引用量达到了800多篇(2020年8月),是引用量最高,收集范围最全面,更新最及时的数据库。vfdb是由中国医学科学院研发,收集整理了多种重要医学病原菌的已知毒素因子的组成、结构、功能、致病机理、毒力岛、序列和基因组信息等内容。
4.目前毒素因子的主要检测方法有两类:(1)根据dna序列设计引物,基于pcr扩增的原理设计成试剂盒,从而对目标dna片段进行扩增,基于荧光强度确认有无;(2)对dna片段进行测序,将测序后的序列做物种和毒素基因的注释分析,从而确定毒素因子的存在。但是方法一通常只能针对几个常见的基因做鉴定和分型,而方法二虽然理论上可以注释出大量的毒素基因,但是只能做到相对丰度的计算,确定毒素因子的存在而无法衡量其强弱。在高通量测序技术的辅助下,宏基因组研究飞速发展,定量宏基因组学也越来越得到重视,定量宏基因组是通过在样本中添加固定量的已知序列作为统一内参,计算样本中每种基因的拷贝数后,基于内参做一致性的标准化,从而实现绝对定量。相比于普通的宏基因组测序技术,定量宏基因组技术能够更准确的反映样本中基因的真实拷贝数和样本组间的真实差异,更准确的反映样本中微生物群落的真实变化,不仅仅具有前沿的科学价值,也具有实际的应用意义。在这里,我们基于定量宏基因组高通量测序技术,检测并且定量了测序样本中毒素基因的丰度,在更广阔的范围和更精确的分辨率上实现了毒素因子的检测。
技术实现要素:
5.为了克服现有技术中的上述不足,本发明的目的之一在于提供一种基于宏基因组学的毒素基因丰度检测方法,可以检测出疾病相关的毒素因子基因,能够更准确的反映样本中基因的真实拷贝数和样本组间的真实差异,更准确的反映样本中微生物群落的真实变化。
6.本发明的目的之二在于提供一种毒素因子基因的注释数据库构建方法,将检测到的毒素基因进行注释,阐释了毒素因子关联的物种和作用的机制。
7.本发明的目的之三在于提供一种计算机可读存储介质。
8.为了实现上述目的之一,本发明采用以下技术方案:
9.本发明提供一种引物探针组合物,包括以下步骤:
10.s1、样本采集并且建库测序,获得待测样本的宏基因组测序数据;
11.s2、对所述宏基因组测序数据进行筛选,得到高质量的测序序列;
12.s3、对所述高质量的测序序列与毒素基因参考数据库进行比对,获取每个基因的比对reads数量;
13.s4、根据基因长度将read数量标准化为相对丰度,然后基于内参基因序列的拷贝数计算每个毒素基因的绝对丰度;
14.其中所述新的毒素因子参考序列数据库是通过添加已知量的内参基因序列到毒素因子参考序列数据库中形成的。
15.进一步地,所述步骤s1中包括步骤:
16.所述样本是采集的待检测用户的粪便组织,提取所述的粪便组织中的菌群dna并且进行质控;
17.向质控合格的所述菌群dna中添加已知量的内参dna,基于小片段文库构建方法建立dna文库并且质控;
18.将质控合格的所述dna文库,进行双端测序,获得所述待测样本的宏基因组测序数据。
19.进一步地,所述步骤s2中包括步骤:
20.在获得所述待测样本的宏基因组测序数据后,去除接头序列以及低质量碱基序列;
21.通过与宿主基因组比对,去除来源于所述宿主基因组的reads,得到所述高质量的测序序列。
22.为了实现上述目的之二,本发明采用以下技术方案:
23.本发明提供一种毒素基因的注释数据库构建方法,包括对毒素因子参考序列数据库进行注释,构建成毒素基因的注释数据库。
24.进一步地,基于所述毒素因子参考序列数据库,将每个所述毒素基因生成其类型、相关物种及作用机制的注释。
25.为了实现上述目的之三,本发明采用以下技术方案:
26.本发明提供一种计算机可读存储介质,所述计算机可读介质能被处理器执行以实现一种基于宏基因组学的毒素基因丰度检测方法及其注释数据库构建方法。
27.与现有技术相比,本发明具有的有益效果如下:
28.(1)本发明提供的一种基于宏基因组学的毒素基因丰度检测方法,可以检测出疾病相关的毒素因子基因,能够更准确的反映样本中基因的真实拷贝数和样本组间的真实差异,更准确的反映样本中微生物群落的真实变化,以在在更广阔的范围和更精确的分辨率上实现了毒素因子的检测。
29.(2)本发明提供的一种毒素因子基因的注释数据库构建方法,基于预建立的新的毒素因子参考序列数据库,将检测到的毒素因子做了注释,阐释了毒素因子关联的物种和作用的机制,可以辅助临床医生判断患者是否具有特定毒素感染,并且这些毒素因子还有
潜力用作治疗靶点,提高病人的治疗效果。
30.(3)本发明的粪便样品是方便运输的,并且粪便采样方法是无创的和舒适的,所以人们将更容易参与到指定过程中。
附图说明
31.为了更清楚地说明本技术实施例中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本技术的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
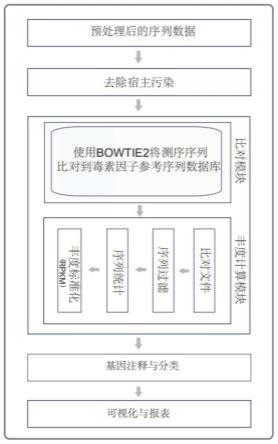
32.图1是本技术实施例1提供的肠道毒素基因丰度检测方法及其注释数据库构建方法的流程示意图。
33.图2是本技术实施例2提供的肠道毒素因子的kegg注释结果示意图。
34.图3是本技术实施例1提供的肠道毒素基因丰度检测方法在模拟数据集中的检测准确性评估。
具体实施方式
35.为了使本技术要解决的技术问题、技术方案及有益效果更加清楚明白,以下将结合实施例对本技术的技术方案进行清楚、完整的描述。应当理解此处所描述的具体实施例仅仅用以解释本技术,并不用于限定本技术。
36.本发明的实施例主要包括采用定量宏基因组测序技术,检测粪便组织中毒素基因的绝对拷贝数,注释相关的分子机制和致病菌,并且筛选常见的毒素基因,构建毒素基因检测试剂盒,可以检测出疾病相关的毒素因子基因,能够更准确的反映样本中基因的真实拷贝数和样本组间的真实差异,更准确的反映样本中微生物群落的真实变化,以在在更广阔的范围和更精确的分辨率上实现了毒素因子的检测;基于预建立的新的毒素因子参考序列数据库,将检测到的毒素因子做了注释,阐释了毒素因子关联的物种和作用的机制,可以辅助临床医生判断患者是否具有特定毒素感染,并且这些毒素因子还有潜力用作治疗靶点,提高病人的治疗效果。
37.实施例1
38.参见附图1,为本发明实施例提供的一种基于宏基因组学的毒素基因丰度检测方法的流程示意图,该方法可以包括以下步骤:
39.s1、样本采集并且建库测序,获得待测样本的宏基因组测序数据。
40.为了研究肠道内毒素基因的特征,募集了1005例志愿者,收集了其粪便样本并且做了定量宏基因组测序,每个样本的测序数据量不低于5g测序序列数据以获得尽可能多的毒素基因信息。
41.在本发明实施例的一种实施方式中,采集检测用户的粪便组织,提取粪便组织中的菌群dna并且进行质控,向质控合格的所述菌群dna中添加已知量的内参dna(spike),基于小片段文库构建方法建立dna文库并且质控,将质控合格的dna文库,进行pair end(pe)双端测序,获得待测样本的宏基因组测序数据。
42.s2、对所述宏基因组测序数据进行筛选,得到高质量的测序序列。
43.具体的,在获得所述待测样本的宏基因组测序数据后,使用了trimmomatic软件对原始的测序数据进行了接头序列的去除以及低质量碱基序列的过滤等数据过滤操作,使用bowtie2将测序序列比对到人类参考基因组上,并且将能够比对上的序列去除掉,以达到的移除宿主污染的目的,得到所述高质量的测序序列。
44.s3、对所述高质量的测序序列与毒素基因参考数据库进行比对,获取每个基因的比对reads数量。
45.具体的,使用bowtie2将所述高质量的测序序列与所述新的毒素基因数据库进行比对,统计比对到每条基因上的唯一比对和多比对双端reads数量;
46.基于所述唯一比对上的reads分布,将所述多比对的reads分配到每个基因上,计算出每个基因的比对reads数量;
47.计算公式如下:
48.公式1:rc(g)=urc(g) mrc(g)
49.公式2:
50.公式3:
51.公式4:
52.其中,rc(read count)代表reads的数目,urc(unique read count)代表唯一比对的reads数目,mrc(multiple aligned read count)代表重复比对的reads数目,fragment表示片段,g代表基因,l(g)表示基因的长度,coj是一个read对第j个基因的贡献权重。
53.s4、根据基因长度将read数量标准化为相对丰度,然后基于内参基因序列的拷贝数计算每个毒素基因的绝对丰度。
54.在获取每个基因的比对reads数量后,根据基因长度将read数量标准化为相对丰度,然后基于内参基因序列的拷贝数计算每个毒素基因的绝对丰度,相对丰度是每千个碱基上的reads数量(rpkm),然后基于内参基因序列的拷贝数计算每个毒素基因的绝对丰度,详细计算方法如下:
55.公式1:rpkmj=coj*109/totalmappedreads
56.其中,coj是一个read对第j个基因的贡献权重,rpkmj表示第j个基因上比对到每千个碱基的reads数量。
57.在本实施例中,需要说明的是为了实现毒素基因的绝对定量,下载vfdb毒素因子数据库,将内参的dna序列加入到vfdb数据库中,形成了新的毒素因子参考序列数据库,为了方便后续的比对分析,在所述使用bowtie2将所述高质量的测序序列与所述毒素基因数据库进行比对之前,使用bowtie2对所述新的毒素因子参考序列数据库做了数据库快速查询索引。
58.本发明的方法可以用来检测用户肠道内毒素因子的分布状况。根据本发明得出的毒素因子检测结果,可以辅助临床医生判断患者是否具有特定毒素感染,并且这些毒素因子还有潜力用作治疗靶点,提高病人的治疗效果。
59.实施例2
60.本发明的另外一方面提供了一种特定毒素因子注释数据库的构建和注释的方法,
61.对每条毒素基因序列做了物种来源和功能分类注释,整合注释信息到毒素因子参考序列数据库中,形成了毒素因子注释数据库,然后通过毒素因子注释数据库对毒素因子进行kegg注释分析,结果如附图2所示。
62.从图2中可以看出,检测到了普遍存在的粘附型、抗吞噬型和分泌型的毒素因子的基因。
63.基于预建立的毒素因子注释数据库,将检测到的毒素因子进一步做了注释,阐释了毒素因子关联的物种,作用的机制等,最终的检测的部分结果如表1所示。
64.表1毒素因子注释结果
[0065][0066]
实验例:
[0067]
数据模拟
[0068]
为了研究该方法检测毒素基因的性能,使用毒素因子参考序列数据库中的dna序列,使用insilicoseq软件模拟了10m的模拟测序序列,并且计算了每种毒素基因理论上的序列数和相对丰度。
[0069]
毒素基因丰度计算
[0070]
使用bowtie2将模拟的测序序列比对毒素基因参考序列,统计了每个毒素基因比对上的序列数量,并且使用rpkm的方法做了进一步的标准化,得到每种毒素基因的相对丰度。
[0071]
算法性能分析
[0072]
为了评估该计算方法的准确度和灵敏度,将理论上的相对丰度与真实丰度进行比较,结果如附图3所示,从图中可以看出,在模拟数据中,正确比对了3010301条序列,有
12600错误分类而12600条序列未能成功比对上,其准确度和召回率分别为99.02%和99.07%。
[0073]
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
