1.本发明属于有机合成领域,具体涉及一种抗组胺药物盐酸非索非那定杂质及其合成方法和应用。
背景技术:
2.盐酸非索非那定是新一代抗组胺药,临床用于各种过敏性疾病,具有很好的耐受性和安全性。该药物具有强大的直接抗炎活性,具有无嗜睡作用、无肝损伤和心脏毒性、副作用很少等临床优势。此外,盐酸非索非那定还是慢性荨麻疹的一线用药,也是抗组胺药物儿童用药的首选。
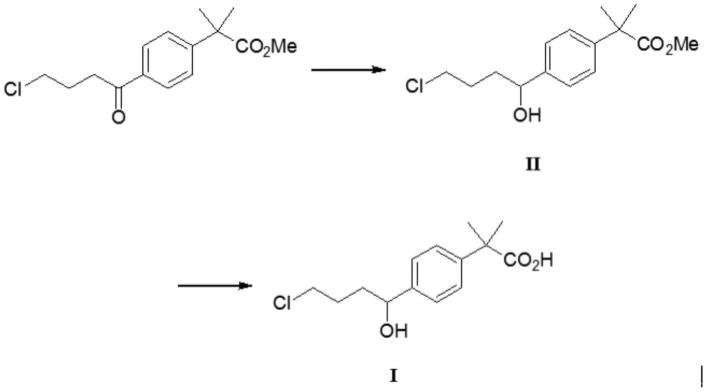
3.在现有技术中,盐酸非索非那定通常采用α,α-二甲基苯乙酸甲酯为原料、经与4-氯丁酰氯进行傅克酰基化获得中间体2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯,再与氮杂环醇进行缩合反应,然后经过还原、水解成盐的方法制备。该合成路线制备过程中会产生一些杂质,这些杂质均已被欧洲药典(european pharmacopoeia 9.0)收录。然而,本技术的发明人在研究中发现,上述方法制备的盐酸非索非那定中间体2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯直接经还原、水解会产生未被药典收录的杂质(化合物i)。
[0004]
随着国家对药物杂质的严格控制以及制剂一致性评价的严格要求,需要建立并完善药物在制备过程中产生的所有杂质的结构信息及图谱信息。
技术实现要素:
[0005]
本技术的发明人在盐酸非索非那定的制备过程中发现,除了现有技术中已经公开的盐酸非索非那定杂质,在采用本领域常规制备方法合成盐酸非索非那定时,还会产生一种如下所示的盐酸非索非那定杂质,
[0006][0007]
对于上述盐酸非索非那定杂质(化合物i),现有技术中未见报道。该杂质的结构相比于原料药盐酸非索非那定缺少了二苯基哌啶基甲醇结构,分子的亲脂成分大大降低了,从而会引起原料药在生物体中的代谢速率减慢,降低药物治疗效果;另一方面该杂质更易在机体产生一定的积累,引起副作用。
[0008]
为了更好的监控盐酸非索非那定原料药合成过程中产生的盐酸非索非那定杂质,本发明提供一种盐酸非索非那定杂质及其合成方法和应用。
[0009]
因此,本发明目的在于提供一种盐酸非索非那定杂质;本发明的另一目的在于提
供上述盐酸非索非那定杂质的合成方法;本发明的又一目的在于提供上述盐酸非索非那定杂质在盐酸非索非那定的质量控制中的应用。
[0010]
实现本发明目的所采取的技术方案如下。
[0011]
一方面,本发明提供一种盐酸非索非那定杂质,所述盐酸非索非那定杂质为如下所示的化合物i:
[0012][0013]
另一方面,本发明提供一种如化合物i所示的盐酸非索非那定杂质的合成方法,
[0014][0015]
所述方法包括如下步骤:
[0016]
(a)选择2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯为原料,甲醇作反应溶剂,在硼氢化钠作用下进行还原反应,降温,调ph,浓缩,析晶,得化合物ii:2-[4-(4-氯-1-羟基)苯基]-2-甲基丙酸甲酯;
[0017]
(b)以步骤(a)获得的化合物ii为原料,采用氢氧化钠水溶液进行水解反应,调ph,降温析晶,得到高纯度的盐酸非索非那定杂质:即化合物i:2-[4-(4-氯-1-羟基)苯基]-2-甲基丙酸。
[0018]
在本发明提供的合成方法中,优选地,在步骤(a)中,所述2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯与硼氢化钠的摩尔比为1:3-5,更优选地为1:4。
[0019]
优选地,在步骤(a)中,所述还原反应的温度为20~50℃,更优选地为20-40℃,最优选地为40℃;
[0020]
优选地,在步骤(a)中,所述还原反应的时间为2小时;
[0021]
优选地,在步骤(a)中,所述降温为降温至0-10℃;优选地为4℃;
[0022]
优选地,在步骤(a)中,所述调ph采用2m稀盐酸溶液进行,更优选地,所述调ph为采用2m稀盐酸溶液将ph调节至中性;
[0023]
优选地,在步骤(a)中,所述析晶按照如下进行:向浓缩得到的浓缩物中加入甲醇和水,于0~10℃下搅拌5-6h析晶;进一步优选地,所述析晶按照如下进行:向浓缩得到的浓缩物中加入体积比为20:9的甲醇和水的混合溶剂,于4℃下搅拌5h析晶。
[0024]
在本发明提供的合成方法中,优选地,在步骤(b)中,所述水解反应的温度为55~
65℃;更优选地为65℃;
[0025]
优选地,在步骤(b)中,水解反应的时间为5小时;
[0026]
优选地,在步骤(b)中,所述调ph采用2m稀盐酸溶液进行,更优选地,所述调ph为采用2m稀盐酸溶液将ph调节至中性;
[0027]
优选地,在步骤(b)中,所述降温析晶为将温度控制在0~10℃,优选4℃析晶;
[0028]
优选地,在步骤(b)中,所述降温析晶的时间4~8小时,更优选地为5-7小时,最优选地为6小时。
[0029]
在本发明的优选实施方案中,提供一种化合物i的合成方法,其中,所述合成方法所采用的合成路线如下:
[0030][0031]
其中,所述制备方法包括如下步骤:
[0032]
(a)选择2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯为原料,甲醇作反应溶剂,在硼氢化钠作用下进行还原反应,其中,所述原料与硼氢化钠的摩尔比为1:4、还原反应的温度为40℃、还原反应的时间为2小时,然后降温至4℃,调ph至中性,浓缩,向浓缩得到的浓缩物中加入体积比为20:9的甲醇和水的混合溶剂,4℃下搅拌5h析晶,得化合物ii:2-[4-(4-氯-1-羟基)苯基]-2-甲基丙酸甲酯;
[0033]
(b)以步骤(a)获得的化合物ii为原料,采用氢氧化钠水溶液于65℃下进行水解反应达5h,调ph至中性,降温至4℃析晶6h,得到高纯度的盐酸非索非那定杂质:即化合物i:2-[4-(4-氯-1-羟基)苯基]-2-甲基丙酸。
[0034]
又一方面,本发明提供上述盐酸非索非那定杂质或按照上述合成方法制备的盐酸非索非那定杂质在盐酸非索非那定的质量控制中的应用。
[0035]
再一方面,本发明提供上述盐酸非索非那定杂质的合成方法在制备盐酸非索非那定杂质对照品中的应用。
[0036]
本发明的有益效果包括:本发明的合成方法所采用的原料简单易得,实验操作简单、高效,合成得到的产品的纯度高,适宜实验室放大生产。通过本发明所提供的合成方法所制备的盐酸非索非那定杂质可用于杂质的定性及定量分析,为盐酸非索非那定原料药的杂质谱分析提供思路,同时提高了原料药质量的用药安全性。此外,本发明还为相关制剂-盐酸非索非那定片的杂质研究提供了参考,为仿制药一致性评价工作建立了合理的杂质分析原理。
附图说明
[0037]
以下,结合附图来详细说明本发明的实施方案,其中:
[0038]
图1显示了盐酸非索非那定杂质(化合物i)的核磁氢谱图。
具体实施方式
[0039]
以下参照具体的实施例来说明本发明。本领域技术人员能够理解,这些实施例仅用于说明本发明,其不以任何方式限制本发明的范围。
[0040]
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的药材原料、试剂材料等,如无特殊说明,均为市售购买产品。其中,部分试剂和设备购买情况如下:
[0041]
1、2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯:西安万隆制药股份有限公司,以2-甲基-2-苯基丙酸甲酯为原料,通过与4-氯丁酰氯发生傅克酰基化反应制备得到;
[0042]
2、硼氢化钠:分析纯,国药集团化学试剂有限公司;
[0043]
3、盐酸:工业级,湖南尔康制药有限公司;
[0044]
4、氢氧化钠:工业级,天津鹏坤化工有限公司;
[0045]
5、三口瓶:250ml,国药集团化学试剂有限公司;
[0046]
6、真空干燥烘箱:dzf-6090,上海一恒科学仪器有限公司;
[0047]
7、循环真空泵:shz-95b,河南省予华仪器有限公司。
[0048]
实施例1
[0049]
(a)向250ml的三口瓶中加入120ml甲醇和2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯20g,然后加入硼氢化钠10.6g,升温至20℃反应2h。反应液降温至4℃,滴加2m稀盐酸溶液,调节ph呈中性。减压浓缩,浓缩残余液中加入100ml甲醇和45ml纯化水,4℃搅拌5h,体系有大量固体析出,抽滤,得湿品,55℃真空干燥干燥获得化合物ii,摩尔收率78.5%。
[0050]
(b)250ml的三口瓶中加入100ml甲醇、化合物ii 15g、和25ml 4m氢氧化钠水溶液。加热升温至55℃反应5h,滴加2m稀盐酸溶液,调节ph呈中性。控温4℃搅拌析晶4h,体系有大量固体析出,抽滤,得湿品,55℃真空干燥干燥获得化合物i,hplc=89.6%,摩尔收率80.9%。
[0051]
制备得到的化合物i的结构确证信息如下:分子式:c
14h19
clo3,分子量:270.75。
[0052]1h nmr(400mhz,dmso-d6),δ:12.29(s,1h),7.31(d,2h),7.29(d,2h),4.78(d,1h),3.80(d,2h),2.54(s,1h),2.26(d,2h),1.94(d,2h),1.65(s,6h)。参见图1。
[0053]
13
c nmr(400mhz,dmso-d6),δ:178.02,144.21,142.05,125.99,79.99,68.24,45.97,40.43,40.02,39.88,39.60,34.65,26.86,26.05.
[0054]
esi-ms(m/z):271.7[m h]
。
[0055]
实施例2
[0056]
本实施例的方法与实施例1中的基本一致,不同的是:步骤(a)中,反应温度为40℃,获得化合物ii 16.1g,收率80.5%;步骤(b)中,反应温度为55℃、析晶时间为7h,获得化合物i11.7g,收率82.1%,hplc=99.0%。
[0057]
实施例3:步骤(a)涉及的工艺参数的筛选试验
[0058]
本实施例的方法与实施例1中的基本一致,具体改变之处参见下表1。
[0059]
表1:步骤(a)影响因素试验
[0060][0061][0062]
实施例4:步骤(b)涉及的工艺参数的筛选试验
[0063]
本实施例的方法与实施例1中的基本一致,具体改变之处参见下表2。
[0064]
表2:步骤(b)影响因素试验
[0065][0066]
实施例5:
[0067]
(a)向250ml的三口瓶中加入120ml甲醇和2-[4-(4-氯-1-丁酰基)苯基]-2-甲基丙酸甲酯20g,然后加入硼氢化钠10.6g,升温至40℃反应2h。反应液降温至4℃,滴加2m稀盐酸溶液,调节ph呈中性,减压浓缩,浓缩残余液中加入100ml甲醇和45ml纯化水,4℃搅拌5h,体系有大量固体析出,抽滤,得湿品,55℃真空干燥干燥获得化合物ii,摩尔收率80.6%。
[0068]
(b)250ml的三口瓶中加入100ml甲醇、化合物ii 15g、和25ml 4m氢氧化钠水溶液。加热升温至65℃反应5h,滴加2m稀盐酸溶液,调节ph呈中性。控温4℃搅拌析晶6h,体系有大量固体析出,抽滤,得湿品,55℃真空干燥干燥获得化合物i,hplc=99.6%,摩尔收率85.2%。
[0069]
上述实施例只为说明本发明的技术方案及特点,其目的在于让医药化工相关研究人员了解本发明的内容并据以实施,并不能以此限制本发明的保护范围,凡根据本发明思路实质所作的等效变化或相关修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。