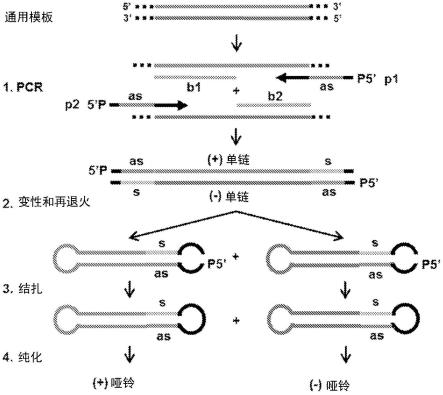
生成哑铃形dna载体的方法
技术领域
1.本公开涉及一种新的通用模板辅助无克隆方法,该方法允许以低成本有效合成哑铃形dna载体,用于将重组dna和rna递送到宿主细胞中。
背景技术:
2.遗传性和获得性遗传疾病的基因治疗、基因疫苗接种、干细胞编程、体细胞重编程、免疫治疗、crispr/cas介导的基因组编辑和体内蛋白质表达操作等方法的效率依赖于将重组dna递送到离体或体内的原代细胞中,以触发非编码rna或蛋白质的表达。
3.在原代细胞中,重组外源游离dna(如质粒)的表达在递送后24小时内被沉默,与递送途径无关。这种影响背后的机制知之甚少。只有整合病毒传递载体,如逆转录病毒、慢病毒和aav载体,已成功用于触发原代细胞的中长期表达。然而,考虑到当前的良好生产规范(cgmp)生产标准,这些载体的成本很高。在cgmp标准下生产病毒载体被认为比生产等量的“裸露”遗传物质要贵几个数量级。此外,病毒载体存在安全风险和担忧,这些风险和担忧与(i)和整合位点的整合外源dna的负面干扰有关(例如破坏基因功能和调控),以及(ii)涉及源自致病病毒的成分。
4.或者,将功能性rna直接递送到原代细胞会导致快速降解并仅提供短期影响。因此,强烈希望开发能够逃避转基因沉默的新型遗传载体。
5.新型载体,如仅由包含启动子、编码基因和rna稳定序列的转录单位组成的dna小环或哑铃形载体,由于体积小而具有若干优点,例如改善细胞递送或核扩散。此外,由于共价闭合结构,这些小载体对核酸外切酶具有抗性,而质粒通常具有由剪切力触发的单链断裂,即所谓的切口。不需要的细菌序列或抗性蛋白的缺乏消除了宿主中不需要的副作用,以及受控的体外合成和将荧光团、细胞穿透肽、抗体、适体、糖或免疫刺激肽化学连接到环结构的选择,允许轻松操作这些载体。
6.如上所述,质粒中的转基因沉默是常见的。在转基因表达盒的5’和3’末端之间缺乏基因外间隔的dna小环被证明允许在小鼠中持续表达转基因。与小圆相比,哑铃形载体的分子量可以小一个数量级,尤其是那些用于表达小非编码rna的载体。wo2012/032114公开了包含哑铃形环状载体的dna表达构建体,该载体在注射入黑素瘤后7天保持表达。与传统载体的生产相比,哑铃形载体的合成通常复杂且成本高。最先进的技术通常依赖于酶,并且还需要克隆和化学合成。尽管已经对方法进行了改进,例如在us2008/0153763中公开的利用基于pcr的技术合成哑铃载体,但这些方法仍然很大程度上依赖于克隆和限制性内切酶,使得哑铃形载体的生产成本很高。
7.micrornas(mirna)代表在大多数后生动物中转录后调节基因表达的小型非编码rna(ncrna)。它们从mirna基因转录为初级mirna转录物(pri-mirna),在被输出到细胞质之前,它们在细胞核中被加工成发夹结构的前体mirna(pre-mirna),在那里它们被进一步加工形成不完美的短配对的rna双链体(mirna:mirna)。小或短发夹rna(shrna)是模拟前mirna的发夹结构的rna,可以从重组基因内源性转录以有效触发rna干扰(rnai)。对于非编
码rna,包括pre-mirna和shrna,以及编码rna基因传递,研究人员探索病毒或非病毒传递载体。虽然病毒载体成本高昂并且经常引发免疫反应或构成基因组载体整合的风险,但许多非病毒递送载体涉及可能对细胞
1,2
有毒的非核酸辅助功能。最简单的非病毒载体是基于裸dna的载体系统,迄今为止已经描述了三种不同类型的载体:质粒、dna小环和哑铃形dna最小载体。虽然基于质粒的基因表达在原代细胞和体内迅速沉默,但小环和哑铃载体不会受到转基因沉默的影响,并且在临床前和临床试验
3-7
中显示出有希望的结果。然而,与由于循环张力8而需要最小尺寸为300个碱基对(bp)的小圆相比,哑铃载体没有下限,并且实际上可以像shrna基因一样短。小哑铃尺寸与其线性结构相结合,可促进细胞递送,尤其是核载体扩散9。已报道了四种方法用于生成表达shrna的哑铃载体:首先,核酸酶辅助的酶促连接(elan),一种方案,其中通过对错误连接的非通路产物进行核酸内切来支持分子间的哑铃连接
10
;其次,一种方案,其中通过pcr扩增表达盒,然后切割酶切割以生成5’突出端(overhang),然后形成分子内连接中的哑铃环
11,12
;第三,一种结合前两个方案的特征的方法,生成尺寸最小的发夹模板转录shrna表达哑铃载体
9,13
;最后,基于空位引物pcr的方法,其采用化学修饰的引物和分子内连接来有效生成具有内部环和改进的核靶向活性的优质哑铃载体
14
。通常,在分子内连接反应期间形成哑铃结构的方案表现出最高的载体产量。为了生成用于表达新的非编码或编码rna的哑铃载体,所有上述方案都依赖于克隆步骤和/或需要核酸内切酶。
8.所公开的方法允许为非编码rna(包括pre-mirna和shrna rna)和编码rna表达生成发夹模板转录哑铃形载体的无克隆生成。它描述了一种基于pcr的方法,该方法使用通用模板,通过pcr引物引入编码特定pre-mirna、shrna、蛋白质或肽的序列。这种新颖的方案生成最小尺寸的发夹模板转录哑铃,不需要任何限制或切口核酸内切酶,并且与高通量兼容。所公开的方法加速并降低了pre-mirna、shrna和编码rna的哑铃载体的生成。所公开的方法允许按照功能基因组学和筛选研究的要求生产高度平行的哑铃载体,并且按照临床前和临床应用的要求,它允许大规模生产哑铃。
9.发明简述
10.根据本发明的一个方面,提供了一种哑铃形表达载体,其中所述载体包括:
11.i)一个或多个线性或发夹形转录盒,每个包含编码待表达的核酸分子的核苷酸序列;
12.ii)可操作地连接到所述转录盒的最小转录启动子核苷酸序列;
13.iii)包含dna核靶向序列的核苷酸序列;
14.iv)核苷酸序列,其包含增强子核苷酸序列和任选地至少一个与所述增强子核苷酸序列相关的内含子,以增强所述表达的核酸分子的表达;
15.v)包含转录后调控元件或组成型核转运元件的核苷酸序列;以及
16.vi)核苷酸序列,其包含与哺乳动物基因组的一部分具有同源性的序列,该序列可用作修复模板,该修复模板是单链或双链的,用于rna引导的基因组编辑。
17.在本发明的一个优选实施方案中,所述最小转录启动子序列进一步包含转录终止核苷酸序列,其中转录起始和终止核苷酸序列可操作地偶联。
18.在本发明的另一优选实施方案中,所述载体包含至少一个内部环结构域。优选地,所述环结构域包含无碱基位点或核苷酸错配。
19.在本发明的一个优选实施方案中,所述无碱基位点包括一个或多个
20.在本发明的一个优选实施方案中,所述无碱基位点包含一个或多个无嘌呤/无嘧啶无碱基位点。
21.在本发明的一个优选实施方案中,所述核苷酸错配包括无碱基位点的基于四氢呋喃的模拟物。
22.在本发明的一个优选实施方案中,所述转录后调节元件是wpre[seq id no 11]。
[0023]
在本发明的一个优选实施方案中,上述i)-vi)中所述的载体核酸分子是单链或双链核酸。
[0024]
在本发明的一个优选实施方案中,所述哺乳动物基因组是人的。
[0025]
在本发明的一个优选实施方案中,所述待表达的核酸分子编码治疗性蛋白质或肽。
[0026]
在本发明的一个优选实施方案中,所述治疗性蛋白质是cas9、cas9n、hspcas9或hspcas9n。
[0027]
在本发明的一个优选实施方案中,所述治疗性蛋白质或肽触发死亡信号。
[0028]
触发细胞死亡信号的蛋白质或肽的例子是本领域已知的。例如,已知细菌毒素如霍乱毒素或白喉毒素、α毒素、炭疽毒素、外毒素、百日咳毒素、志贺毒素、志贺样毒素等会诱导细胞死亡。此外,凋亡信号/蛋白质,如fas、tnf、半胱天冬酶(起始半胱天冬酶,半胱天冬酶2、8、9、10、11、12和效应半胱天冬酶,半胱天冬酶3、6、7)等。将无毒药物转化为有毒成分:例如单纯疱疹病毒胸苷激酶(hsvtk)将相当无毒的药物更昔洛韦(gcv)转化为有毒的三磷酸盐(hsvtk/gcv系统)。另一例子是大肠杆菌嘌呤核苷磷酸化酶(pnp)/氟达拉滨自杀基因系统。
[0029]
在本发明的另一优选实施方案中,所述治疗性蛋白质或肽是hsvtk。
[0030]
在本发明的另一优选实施方案中,所述表达的核酸分子是治疗性核酸分子。
[0031]
在本发明的一个优选实施方案中,所述治疗性核酸是sirna或shrna。
[0032]
在本发明的另一优选实施方案中,所述治疗性核酸分子是反义rna寡核苷酸或反义mirna。
[0033]
在本发明的另一优选实施方案中,所述治疗性核酸分子是mirna。
[0034]
在本发明的另一优选实施方案中,所述治疗性核酸分子是反式剪接rna。
[0035]
在本发的另一优选的实施方案中,所述治疗性核酸分子是引导rna、单引导rna、crrna或tracrrna。
[0036]
在本发明的一个优选实施方案中,所述治疗性核酸分子是转拼rna。
[0037]
在本发明的另一优选实施方案中,所述治疗性核酸分子是pre-mrna或mrna。
[0038]
在本发明的另一优选实施方案中,所述最小转录启动子源自rna聚合酶iii启动子。
[0039]
在本发明的一个优选实施方案中,所述rna聚合酶iii启动子是u6启动子并且包含如seq id no:1所示的核苷酸序列。
[0040]
在本发明的另一优选实施方案中,所述rna聚合酶iii启动子是包含如seq id no:2所示核苷酸序列的h1启动子。
[0041]
在本发明的另一优选实施方案中,所述rna聚合酶iii启动子是最小h1(mh1)启动
子,其包含如seq id no:3所示的核苷酸序列。
[0042]
在本发明的另一备选优选实施方案中,所述rna聚合酶iii启动子是修饰的mh1启动子,其包括限制性核酸内切酶切割位点和/或包含如seq id no:4所示核苷酸序列的反向聚合酶iii转录终止子。
[0043]
在本发明的另一优选实施方案中,所述最小转录启动子源自rna聚合酶ii启动子。
[0044]
在本发明的一个优选实施方案中,所述rna聚合酶ii启动子是cmv启动子并且包含如seq id no:5所示的核苷酸序列。
[0045]
在本发明的一个优选实施方案中,所述转录终止子核苷酸序列是rna聚合酶ii或rna聚合酶iii终止序列。
[0046]
在本发明的一个优选实施方案中,所述rna聚合酶iii终止序列包含一个或多个包含核苷酸序列ttttt的基序。
[0047]
在本发明的一个优选实施方案中,所述dna核靶向序列包含在seq id no:6(dtsα和/或seq id no:7(dtsβ)中列出的核苷酸序列。
[0048]
在本发明的一个优选实施方案中,所述增强子核苷酸序列包含在seq id no:8中列出的核苷酸序列(最小增强子:msv40enh)。
[0049]
在本发明的另一优选实施方案中,所述增强子核苷酸序列包含在seq id no:9中列出的核苷酸序列(全长增强子:fsv40enh)。
[0050]
在本发明的另一优选实施方案中,所述内含子包含在seq id no:10中列出的核苷酸序列。
[0051]
在本发明的另一优选实施方案中,所述载体进一步编码可检测标记。
[0052]
在本发明的一个优选实施方案中,所述可检测标记是荧光标记。
[0053]
在本发明的一个优选实施方案中,所述荧光标记是荧光报告蛋白。
[0054]
通过将启动子与编码“报告”蛋白质或多肽的核酸融合,可以方便地监测组织中启动子活性的分析。实例在本领域中是众所周知的并且包括酶如β葡糖苷酸酶。作为蛋白质荧光团的报告分子也是本领域已知的。绿色荧光蛋白gfp是一种从腔肠动物(如太平洋水母、维多利亚水母)中分离出来的自发荧光蛋白。它的作用是通过能量转移将另一种蛋白质水母发光蛋白的蓝色化学发光转换为绿色荧光。gfp可以作为蛋白质标签发挥作用,因为它可以耐受n端和c端与多种蛋白质的融合,其中许多蛋白质已被证明保留了天然功能。大多数情况下,它以增强型gfp的形式使用,其中密码子的使用适应人类密码。其他蛋白质荧光团包括黄色、红色和蓝色荧光蛋白。这些可从例如clontech(www.clontech.com)商购获得。另一例子是萤火虫萤光素酶。
[0055]
在本发明的一个优选实施方案中,其中与哺乳动物基因组的一部分具有同源性的所述核苷酸序列被实施到哑铃载体的双链dna部分中。
[0056]
在本发明的另一优选实施方案中,与哺乳动物基因组的一部分具有同源性的所述核苷酸序列包含哑铃载体的单链环。
[0057]
根据本发明的另一方面,提供了包含根据本发明的哑铃形载体的药物组合物。
[0058]
本发明的哑铃形载体组合物以药学上可接受的制剂施用。这种制剂通常可以含有药学上可接受浓度的盐、缓冲剂、防腐剂、相容的载体和补充治疗剂。本发明的哑铃形载体组合物可以通过任何常规途径施用,包括注射或随时间逐渐输注。施用可以以例如静脉内、
腹膜内、肌内、腔内、皮下、经皮、口服、局部、气管内、鼻内、阴道内或经上皮等方式。或者,本发明的哑铃形载体或载体组合物通过物理方法递送,包括但不限于液体喷射注射、显微注射、微针、粉末颗粒注射、金颗粒注射、基因枪、电穿孔或流体动力注射。
[0059]
本发明的哑铃形载体组合物以有效量施用。“有效量”是单独或与其他剂量一起生成所需反应的哑铃形载体的量。在治疗疾病的情况下,所需的反应是抑制疾病的进展。这可能仅涉及暂时减缓疾病的进展,尽管更优选地,它涉及永久停止疾病的进展。这可以通过常规方法监测。当然,这样的量将取决于正在治疗的特定病症、病症的严重程度、个体患者参数,包括年龄、身体状况、体型和体重、治疗持续时间、同时治疗的性质(如果有)、具体的施用途径和健康从业者的知识和专长范围内的类似因素。这些因素对于本领域普通技术人员来说是众所周知的并且可以通过不超过常规实验的范围来解决。通常优选使用单个组分或其组合的最大剂量,即根据合理的医学判断的最高安全剂量。然而,本领域普通技术人员将理解,患者可能出于医学原因、心理原因或几乎任何其他原因坚持较低剂量或可耐受剂量。
[0060]
上述方法中使用的哑铃形载体组合物优选是无菌的,并且含有有效量的根据本发明的哑铃形载体,用于以适合施用于患者的重量或体积单位生成所需的反应。可以根据不同的参数,特别是根据使用的施用方式和对象的状态来选择施用对象的载体剂量。其他因素包括所需的治疗时间。如果在应用的初始剂量下对象的反应不充分,则可以在患者耐受允许的范围内使用更高剂量(或通过不同的、更局部的递送途径有效地更高剂量)。本领域普通技术人员将已知用于施用载体组合物的其他方案,其中剂量、注射时间表、注射部位、施用方式等与前述不同。在与上述基本相同的条件下进行组合物对人类以外的哺乳动物的施用(例如,用于测试目的或兽医治疗目的)。如本文所用,对象是哺乳动物,优选是人,并且包括非人灵长类动物、牛、马、猪、绵羊、山羊、狗、猫或啮齿动物。
[0061]
当施用时,本发明的哑铃形载体组合物以药学上可接受的量和药学上可接受的组合物施用。术语“药学上可接受的”是指不干扰活性剂的生物活性的有效性的无毒材料。此类制剂通常可包含盐、缓冲剂、防腐剂、相容载体和任选的其他治疗剂(例如,通常用于治疗特定疾病适应症的治疗剂)。当用于药物时,盐应该是药学上可接受的,但非药学上可接受的盐可以方便地用于制备其药学上可接受的盐并且不排除在本发明的范围之外。此类药理学和药学上可接受的盐包括但不限于由以下酸制备的那些:盐酸、氢溴酸、硫酸、硝酸、磷酸、马来酸、乙酸、水杨酸、柠檬酸、甲酸、丙二酸、琥珀酸等。此外,药学上可接受的盐可以制备成碱金属盐或碱土盐,例如钠盐、钾盐或钙盐。
[0062]
根据本发明的含有哑铃形载体的药物组合物可以含有合适的缓冲剂,包括:盐中的乙酸;盐中的柠檬酸;盐中的硼酸;和磷酸盐。药物组合物还可以任选含有合适的防腐剂,例如:苯扎氯铵;氯丁醇;对羟基苯甲酸酯和硫柳汞。
[0063]
哑铃形载体组合物可以方便地以单位剂型存在并且可以通过药学领域中众所周知的任何方法制备。所有方法都包括使活性剂与构成一种或多种辅助成分的载体结合的步骤。含有根据本发明的载体的组合物可以作为气雾剂和吸入施用。适用于肠胃外施用的组合物方便地包含载体的无菌水性或非水性制剂,其优选与接受者的血液等渗。该制剂可以根据已知方法使用合适的分散剂或润湿剂和悬浮剂来配制。无菌可注射制剂也可以是在无毒的肠胃外可接受的稀释剂或溶剂中的无菌可注射溶液或悬浮液,例如作为在1,3-丁二醇中的溶液。可接受的溶剂包括水、林格溶液和等渗氯化钠溶液。此外,无菌的不挥发油通常
用作溶剂或悬浮介质。为此目的,可以使用任何温和的不挥发油,包括合成的甘油单酯或甘油二酯。此外,脂肪酸如油酸可用于制备注射剂。适用于口服、皮下、静脉内、肌肉内等施用的载体制剂可以在remington’s pharmaceutical sciences,mack publishing co.,easton,pa中找到。
[0064]
在本发明的另一实施方案中,所述药物组合物是包含佐剂和/或载体的dna疫苗组合物。
[0065]
根据本发明的一个方面,提供了一种生成包含两个表达盒的哑铃形双表达载体的方法,其中所述载体包括:
[0066]
i)提供包含包含靶核酸分子的第一单链核酸模板的制剂,该靶核酸分子包含在5’末端序列处的第一转录启动子的反向互补序列和在3’末端序列处的第二转录终止子的序列;
[0067]
ii)使所述第一单链核酸模板与第一寡核苷酸引物接触,其包含与所述单链核酸模板的3’末端核苷酸序列的至少部分互补的5
’‑
磷酸和3
’‑
羟基,并且进一步包含与靶核酸分子不互补的5’核苷酸序列,其中,所述寡核苷酸引物包含待转录的第一感兴趣的序列,进一步包含第一转录终止子的反向互补序列,并且进一步包含修饰的核苷酸序列,其防止与靶核酸分子不互补的5’核苷酸序列的延伸;
[0068]
iii)提供聚合酶链式反应组分和引物延伸3’退火的寡核苷酸引物以形成第二模板;
[0069]
iv)使所述第二模板与第二寡核苷酸引物接触,其包含与所述第二模板的3’末端核苷酸序列的至少部分互补的5
’‑
磷酸和3
’‑
羟基,并且进一步包含与第二模板不互补的5’核苷酸序列,其中,所述寡核苷酸引物包含待转录的第二感兴趣的序列,并且进一步包含第二转录终止子的反向互补序列,并且进一步包含防止与第二模板不互补的5’核苷酸序列延伸的修饰的核苷酸序列;
[0070]
v)提供聚合酶链式反应组分和引物延伸3’退火的寡核苷酸引物以形成双链核酸;
[0071]
vi)聚合酶链扩增双链核酸以合成模板dna池并将所述模板退火以生成包含与靶核酸分子不互补的5’核苷酸序列的双链核酸;以及
[0072]
vii)使退火的模板核酸与5dna连接酶接触以将不互补5’核苷酸序列的末端5
’‑
磷酸连接到所述扩增的模板核酸的3
’‑
oh以生成末端环结构。
[0073]
在本发明的优选方法中,所述靶核苷酸分子在其3’末端核苷酸序列处包含转录启动子的序列,并且在其5’末端核苷酸序列处进一步包含转录启动子的反向互补序列。
[0074]
在另一优选的方法中,所述转录启动子是聚合酶iii启动子。
[0075]
在另一优选的方法中,所述聚合酶iii启动子是u6启动子。
[0076]
在另一优选的方法中,所述聚合酶iii启动子是h1启动子。
[0077]
在另一优选的方法中,所述聚合酶iii启动子是最小的h1启动子。
[0078]
在另一种优选的方法中,所述转录启动子是聚合酶ii启动子。
[0079]
在另一优选的方法中,所述聚合酶ii启动子是cmv启动子。
[0080]
在本发明的优选方法中,所述寡核苷酸引物包含与所述靶核酸分子不互补但在其部分长度上包括形成茎环结构的内部互补区域的核苷酸序列。
[0081]
在本发明的优选方法中,所述寡核苷酸引物在其部分长度上包括回文核苷酸序
列。
[0082]
在本发明的优选方法中,所述寡核苷酸引物修饰包括在所述引物中的dna聚合酶引物延伸期间不被识别为碱基配对模板的位点。
[0083]
在本发明的优选方法中,所述寡核苷酸引物修饰是在所述引物中包含无碱基位点。
[0084]
无碱基位点通常由dna损伤或通过自发突变引起,并在dna或rna中定义一个既没有嘌呤也没有嘧啶碱基的位置。这些位点被称为无嘌呤或无嘧啶。
[0085]
在另一优选的方法中,所述无碱基位点是无嘌呤/无嘧啶位点。
[0086]
在另一优选的方法中,所述无嘌呤/无嘧啶位点包括四氢呋喃。
[0087]
在另一优选的方法中,所述无碱基位点包含至少一个或至少三个无嘌呤/无嘧啶位点。
[0088]
在另一优选的方法中,所述无碱基位点包含一个无嘌呤/无嘧啶位点。
[0089]
在本发明的另一优选的方法中,所述无碱基位点将与所述单链核酸模板的3’末端核苷酸序列互补的区域和与靶核酸分子不互补的5’核苷酸序列分开。
[0090]
在本发明的优选方法中,所述寡核苷酸引物包含不互补核苷酸序列,所述核苷酸序列包含转录终止子。
[0091]
在本发明的另一优选的方法中,所述转录终止子聚合酶iii转录终止子。
[0092]
在另一优选的方法中,所述聚合酶iii转录终止子是小的聚胸苷(t)延伸(stretch)。
[0093]
在另一优选的方法中,所述聚合酶iii转录终止子是t五聚体。
[0094]
在本发明的另一优选方法中,所述转录终止子是聚合酶ii转录终止子。
[0095]
在另一优选的方法中,所述聚合酶ii转录终止子是sv40多腺苷酸化位点。
[0096]
在本发明的优选方法中,所述寡核苷酸引物包含待转录的感兴趣序列。
[0097]
在本发明的另一优选的方法中,所述感兴趣的序列是非蛋白质编码序列。
[0098]
在本发明的另一种优选方法中,所述感兴趣的序列是蛋白质编码序列。
[0099]
在本发明的优选方法中,所述dna连接酶是噬菌体dna连接酶,例如t4 dna连接酶或大肠杆菌dna连接酶。
[0100]
在本发明的另一优选方法中,所述dna连接酶是环连接酶。
[0101]
根据本发明的一个方面,提供了一种无克隆和无核酸内切酶的方法来生成包含发夹结构表达盒的哑铃形载体,包括:
[0102]
i)提供包含单链核酸模板的制剂,所述单链核酸模板包含靶核酸分子,所述靶核酸分子包含两个互补序列区段,其中每个区段包含转录启动子序列并且进一步包含转录终止子,并且其中所述两个互补序列区段由第三序列区段隔开;
[0103]
ii)提供其中所述单链核酸模板额外包含茎的制剂;
[0104]
iii)使所述单链核酸模板与包含3
’‑
羟基的第一寡核苷酸引物接触,所述第一寡核苷酸引物与所述单链核酸模板的3’末端核苷酸序列的至少部分互补,并且进一步包含与所述靶核酸分子不互补的5’核苷酸序列,其中所述寡核苷酸引物包含编码发夹结构rna的3’臂的序列;并且进一步使所述单链核酸模板与第一阻断寡核苷酸接触,所述第一阻断寡核苷酸与所述单链核酸模板的5’末端核苷酸序列的至少部分互补;
[0105]
iv)提供聚合酶链反应组分以对3’退火的第一寡核苷酸引物进行引物延伸;
[0106]
v)使所述延伸的寡核苷酸引物与包含3
’‑
羟基的第二寡核苷酸引物接触,所述第二寡核苷酸引物与所述延伸的寡核苷酸引物的3’末端核苷酸序列的至少部分互补并且进一步包含与所述靶核酸分子不互补的5’核苷酸序列,其中所述寡核苷酸引物包含编码发夹结构rna的3’臂的序列;并且进一步使所述单链核酸模板与第二阻断寡核苷酸接触,所述第二阻断寡核苷酸与所述延伸的寡核苷酸引物的5’末端核苷酸序列的至少部分互补;
[0107]
vi)将所述模板进行聚合酶链扩增以合成模板dna的池并将所述模板退火以生成双链核酸,所述双链核酸在正链和负链的5’核苷酸序列处包含编码发夹结构rna的3’臂的序列,并且在正链和负链的3’核苷酸序列处包含编码发夹结构rna的5’臂的序列;
[0108]
vii)使所述双链核酸热变性,然后冷却以允许所得正链和负链dna的分子内重新折叠,以建立由正链或负链dna组成的预形成的寡聚茎环结构;
[0109]
viii)将正链和负链dna的所述预形成的寡聚环结构与单链特异性dna连接酶接触以在分子内连接中将末端5
’‑
磷酸化的5’突出端与相同dna链的3’突出端的末端3
’‑
oh基团连接,以建立共价闭合的正链衍生和负链衍生的哑铃形载体dna,其包含发夹结构的模板dna,用于发夹结构rna的转录;
[0110]
ix)将所述哑铃形载体dna与dna核酸外切酶接触,以去除所有引物和含有5’和3’端的非共价闭合dna。
[0111]
在优选的方法中,所述转录启动子是聚合酶iii启动子。
[0112]
在另一优选的方法中,所述聚合酶iii启动子是u6启动子。
[0113]
在另一优选的方法中,所述聚合酶iii启动子是h1启动子。
[0114]
在另一优选的方法中,所述聚合酶iii启动子是最小的h1启动子。
[0115]
在另一优选的方法中,所述转录启动子是聚合酶ii启动子。
[0116]
在另一优选的方法中,所述聚合酶ii启动子是cmv启动子。
[0117]
在优选的方法中,所述转录终止子是聚合酶iii转录终止子。
[0118]
在另一优选的方法中,所述聚合酶iii转录终止子是小的聚胸苷(t)片段。
[0119]
在另一优选的方法中,所述聚合酶iii转录终止子是t五聚体。
[0120]
在另一种优选的方法中,所述转录终止子是聚合酶ii转录终止子。
[0121]
在另一优选的方法中,所述聚合酶ii转录终止子是sv40多腺苷酸化位点。
[0122]
在优选的方法中,所述茎是人造茎。
[0123]
在另一优选的方法中,所述茎是microrna(mirna)茎。
[0124]
在另一优选的方法中,所述茎是mirna茎是mirna-30(mir-30)茎。
[0125]
在优选的方法中,所述第三序列片段是寡聚dna序列。
[0126]
在另一优选的方法中,所述第三序列片段是四聚体dna序列。
[0127]
在另一优选的方法中,所述第三序列片段是t四聚体。
[0128]
在优选的方法中,所述第一寡核苷酸引物包含5’磷酸。
[0129]
在优选的方法中,所述发夹结构的rna是小发夹rna(shrna)。
[0130]
在另一优选的方法中,所述发夹结构的rna是前体mirna(pre-mirna)。
[0131]
在优选的方法中,所述第一阻断寡核苷酸与所述单链核酸模板的一半的完整5’末端核苷酸序列互补。
[0132]
在优选的方法中,所述第二寡核苷酸引物包含5’磷酸。
[0133]
在优选的方法中,所述第二阻断寡核苷酸与所述延伸的寡核苷酸引物的完整5’末端核苷酸序列的一半互补。
[0134]
在优选的方法中,所述热变性包括在96℃孵育5分钟。
[0135]
在优选的方法中,所述冷却包括从96℃逐渐冷却至室温。
[0136]
在本发明的优选方法中,所述连接酶是单链dna特异性连接酶。
[0137]
在本发明的另一优选的方法中,所述连接酶是环状连接酶。
[0138]
在本发明的优选方法中,所述核酸外切酶是t7 dna聚合酶。
[0139]
根据本发明的一个方面,提供了一种无克隆和无核酸内切酶的方法来生成哑铃形载体,所述载体包括表达盒,所述方法包括:
[0140]
i)提供包含单链核酸模板的制剂,所述单链核酸模板包含靶核酸分子,所述靶核酸分子包含两个互补序列区段,其中每个区段包含转录启动子序列并且进一步包含转录终止子,并且其中所述两个互补序列区段由第三序列区段隔开;
[0141]
ii)提供其中所述单链核酸模板额外包含茎的制剂;
[0142]
iii)使所述单链核酸模板与包含3
’‑
羟基的第一寡核苷酸引物接触,所述第一寡核苷酸引物与所述单链核酸模板的3’末端核苷酸序列的至少部分互补,并且进一步包含与所述靶核酸分子不互补的5’核苷酸序列;并且进一步使所述单链核酸与第一组阻断寡核苷酸接触,所述第一组阻断寡核苷酸包含与所述单链核酸模板的5’末端核苷酸序列的至少部分互补的一种、两种、三种或更多种寡核苷酸;
[0143]
iv)提供聚合酶链反应组分以对3’退火的第一寡核苷酸引物进行引物延伸;
[0144]
v)使所述延伸的寡核苷酸引物与包含3
’‑
羟基的第二寡核苷酸引物接触,所述第二寡核苷酸引物与所述延伸的寡核苷酸引物的3’末端核苷酸序列的至少部分互补并且进一步包含与所述靶核酸分子不互补的5’核苷酸序列;并且进一步使所述单链核酸模板与第二组阻断寡核苷酸接触,所述第二组阻断寡核苷酸包含与所述延伸的寡核苷酸引物的5’末端核苷酸序列的至少部分互补的一种、两种、三种或更多种寡核苷酸;
[0145]
vi)将模板进行聚合酶链扩增以合成延伸的寡核苷酸引物的池(并且将所述模板进行退火以建立双链核酸,所述双链核酸在正链和负链的5’核苷酸序列处包含编码发夹结构rna的3’臂的序列,并且在正链和负链的3’核苷酸序列处包含编码发夹结构rna的5’臂的序列);
[0146]
vii)使所述延伸的寡核苷酸引物的池与包含3’羟基的第三寡核苷酸引物接触,所述第三寡核苷酸引物与由所述第一寡核苷酸引物延伸的寡核苷酸引物的3’末端核苷酸序列的至少部分互补,并且进一步包含与所述延伸的寡核苷酸引物不互补的5’核苷酸序列;并且进一步使所述延伸的寡核苷酸引物的池与包含3’羟基的第四寡核苷酸引物接触,所述第四寡核苷酸引物与由所述第二寡核苷酸引物延伸的寡核苷酸引物的3’末端核苷酸序列的至少部分互补,并且进一步包含与所述延伸的寡核苷酸引物不互补的5’核苷酸序列;并且进一步使所述延伸的寡核苷酸引物与所述第一组和第二组阻断寡核苷酸接触;
[0147]
viii)使用第五和第六、第七和八对或更多对寡核苷酸引物重复步骤iv)和vii),其中最后一组引物包含3’羟基基团和5’磷酸基团;
[0148]
ix)将所述模板进行聚合酶链扩增以合成延伸的寡核苷酸引物的池并且将所述模
板退火以建立双链核酸,所述双链核酸在正链和负链的5’核苷酸序列处包含编码发夹结构rna的3’臂的序列,并且在正链和负链的3’核苷酸序列处包含编码发夹结构rna的5’臂的序列;
[0149]
x)使所述双链核酸热变性,然后冷却以允许所得正链和负链dna的分子内重新折叠,以建立由正链或负链dna组成的预形成的寡聚茎环结构;
[0150]
xi)将所述预形成的寡聚环结构的正链和负链dna与单链特异性dna连接酶接触,以分子内连接将末端5
’‑
磷酸化的5’突出端与相同dna链的3’突出端的末端3
’‑
oh基团连接,以建立共价闭合的正链衍生和负链衍生的哑铃形载体dna,其包含发夹结构的模板dna,用于发夹结构rna的转录;
[0151]
xii)将所述哑铃形载体dna与dna核酸外切酶接触,以去除所有引物和含有5’和3’端的非共价闭合dna。
[0152]
在优选的方法中,所述转录启动子是聚合酶iii启动子。
[0153]
在另一优选的方法中,所述聚合酶iii启动子是u6启动子。
[0154]
在另一优选的方法中,所述聚合酶iii启动子是h1启动子。
[0155]
在另一优选的方法中,所述聚合酶iii启动子是最小的h1启动子。
[0156]
在另一优选的方法中,所述转录启动子是聚合酶ii启动子。
[0157]
在另一优选的方法中,所述聚合酶ii启动子是cmv启动子。
[0158]
在优选的方法中,所述茎是人造茎。
[0159]
在另一优选的方法中,所述茎是microrna(mirna)茎。
[0160]
在另一优选的方法中,所述茎是mirna茎是mirna-30(mir-30)茎。
[0161]
在优选的方法中,所述第三序列片段是寡聚dna序列。
[0162]
在另一优选的方法中,所述第三序列片段是四聚体dna序列。
[0163]
在另一优选的方法中,所述第三序列片段是t四聚体。
[0164]
在另一优选的方法中,所述第一和所述第二寡核苷酸引物在它们的5’核苷酸序列中包含翻译起始密码子cat的反向互补序列,与靶核酸分子不互补。
[0165]
在另一优选的方法中,所述最后一组引物的引物在其5’核苷酸序列中包含与所述延伸的寡核苷酸引物不互补的翻译终止密码子的反向互补序列。
[0166]
在另一优选的方法中,所述翻译终止密码子是tag、taa或tga。
[0167]
在备选的优选方法中,所述最后一组引物的引物在其5’核苷酸序列中包含转录终止子的反向互补序列,所述核苷酸序列与所述延伸的寡核苷酸引物不互补。
[0168]
在优选的方法中,所述转录终止子是聚合酶iii转录终止子。
[0169]
在另一优选的方法中,所述聚合酶iii转录终止子是小的聚胸苷(t)片段。
[0170]
在另一优选的方法中,所述聚合酶iii转录终止子是t五聚体。
[0171]
在备选的优选方法中,所述转录终止子是聚合酶ii转录终止子。
[0172]
在另一优选的方法中,所述聚合酶ii转录终止子是sv40多腺苷酸化位点。
[0173]
在另一种优选方法中,所述第一寡核苷酸引物包含5’磷酸。
[0174]
在优选的方法中,所述发夹结构的rna是小发夹rna(shrna)。
[0175]
在另一优选的方法中,所述发夹结构的rna是前体mirna(pre-mirna)。
[0176]
在优选的方法中,所述第一阻断寡核苷酸与所述单链核酸模板的一半的完整5’末
端核苷酸序列互补。
[0177]
在优选的方法中,所述第二寡核苷酸引物包含5’磷酸。
[0178]
在优选的方法中,所述第二阻断寡核苷酸与所述延伸寡核苷酸引物的完整5’末端核苷酸序列的一半互补。
[0179]
在优选的方法中,所述热变性包括在96℃孵育5分钟。
[0180]
在优选的方法中,所述冷却包括从96℃逐渐冷却至室温。
[0181]
在本发明的优选方法中,所述连接酶是单链dna特异性连接酶。在本发明的另一优选的方法中,所述连接酶是环化连接酶(circligase)。
[0182]
在本发明的优选方法中,所述核酸外切酶是t7 dna聚合酶。
[0183]
根据本发明的一个方面,提供了一种通过根据本发明的方法合成的哑铃形载体。
[0184]
根据本发明的另一方面,提供了用于转染分离来自人类对象的原代细胞的方法,包括:
[0185]
i)提供包含待转染细胞的分离样本;
[0186]
ii)形成包含所述分离细胞样本的制剂,并使所述样本与根据本发明的方法制备的哑铃形载体接触;
[0187]
iii)提供能够将所述哑铃形载体引入所述原代细胞样本并持续表达包含在所述载体中的核酸分子的转化条件。
[0188]
根据本发明的另一方面,提供了一种治疗患有将受益于基因治疗的疾病的患者的离体方法,包括以下步骤:
[0189]
i)从所述对象获得包含待转染细胞的样本;
[0190]
ii)形成根据本发明的方法制备的哑铃形载体的细胞培养制剂,并提供将所述载体转染到所述细胞中的条件;以及
[0191]
iii)将转染的细胞施用所述对象。
[0192]
在本发明的优选方法中,所述分离样本包含干细胞。
[0193]
在本发明的一个优选实施方案中,所述干细胞选自:多能干细胞,例如胚胎干细胞或诱导的多能干细胞、多能干细胞、谱系限制性干细胞。
[0194]
术语“干细胞”代表一组未分化细胞,它们具有自我更新的能力,同时保留了形成分化细胞和组织的不同潜力。干细胞可以是多能的或多能的。多能干细胞是具有形成在完整有机体中发现的所有组织的能力的细胞,尽管多能干细胞不能形成完整有机体。多能细胞形成分化细胞和组织的能力有限。通常,成体干细胞是多能干细胞,是前体干细胞或谱系受限的干细胞,具有形成一些细胞或组织并补充衰老或受损细胞/组织的能力。多能干细胞的例子包括间充质干细胞。间充质干细胞或msc分化成多种细胞类型,包括成骨细胞、软骨细胞、肌细胞、脂肪细胞和神经元。通常,msc是从骨髓中获得的,但也可以来自其他来源,例如脂肪组织。
[0195]
在本发明的优选方法中,所述细胞是外周血单核细胞。
[0196]
在本发明的优选方法中,所述外周血单核细胞包括:t淋巴细胞,[cd8
t淋巴细胞或cd4
t淋巴细胞中的一种或两种]b淋巴细胞,树突细胞,t调节细胞,先天性淋巴细胞或自然杀伤细胞[nk细胞]。
[0197]
显然,“外周血单核细胞”可以从血液以外的来源分离,例如淋巴结和脾脏,并且提
及外周血单核细胞并不将本发明限制为从血液中分离的那些细胞。
[0198]
根据本发明的另一方面,提供了一种试剂盒,该试剂盒包括:寡核苷酸引物,其设计为与单链靶核酸模板的3’末端核苷酸序列的至少部分互补,并且进一步包含与靶核酸分子不互补的5’核苷酸序列,其中所述寡核苷酸引物包含修饰的核苷酸序列,当与靶标退火时,该修饰的核苷酸序列防止与靶核酸分子不互补的5’核苷酸序列的延伸。
[0199]
在本发明的一个优选实施方案中,所述试剂盒进一步包含聚合酶链式反应组分。
[0200]
在本发明的一个优选实施方案中,所述试剂盒包含:5热稳定的dna聚合酶、脱氧核苷酸三磷酸和聚合酶链扩增所需的辅助因子。
[0201]
在本发明的一个优选实施方案中,所述试剂盒包括dna连接酶。
[0202]
在本发明的一个优选实施方案中,所述试剂盒进一步包含用于转染细胞的细胞转染组分,优选哺乳动物细胞如人类细胞。
[0203]
发明详述
[0204]
治疗性蛋白质和肽
[0205]
本发明包括哑铃形载体,其包含编码药物蛋白如“细胞因子”的核酸。细胞因子涉及多种不同的细胞功能。这些包括调节免疫系统、调节能量代谢和控制生长发育。细胞因子通过在靶细胞表面表达的受体介导它们的作用。细胞因子的实例包括白细胞介素,例如:il1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32和33。其他例子包括生长激素、瘦素、促红细胞生成素、催乳素、肿瘤坏死因子[tnf]、粒细胞集落刺激因子(gcsf)、粒细胞巨噬细胞集落刺激因子(gmcsf)、睫状神经营养因子(cntf)、心肌营养因子-1(ct-1)、白血病抑制因子(lif)和制瘤素m(osm)、干扰素α、干扰素β、干扰素ε、干扰素κ和ω干扰素。
[0206]
药物活性肽的例子包括glp-1、抗利尿激素、催产素、促性腺激素释放激素、促肾上腺皮质激素释放激素;降钙素、胰高血糖素、胰淀素、a型利钠激素、b型利钠激素、生长素释放肽、神经肽y、神经肽yy
3-36
、生长激素释放激素、生长抑素,或其同源物或类似物。
[0207]
术语“趋化因子”是指由具有促有丝分裂、趋化或炎症活性的细胞分泌的一组结构相关的低分子量因子。它们主要是具有70到100个氨基酸残基的阳离子蛋白,共有四个保守的半胱氨酸残基。这些蛋白质可以根据两个氨基末端半胱氨酸的间距分为两组。在第一组中,两个半胱氨酸被一个残基(c-x-c)隔开,而在第二组中,它们是相邻的(c-c)。“c-x-c”趋化因子成员的例子包括但不限于血小板因子4(pf4)、血小板碱性蛋白(pbp)、白细胞介素8(il-8)、黑色素瘤生长刺激活性蛋白(mgsa)、巨噬细胞炎症蛋白2(mip-2)、小鼠mig(m119)、鸡9e3(或pcef-4)、猪肺泡巨噬细胞趋化因子i和ii(amcf-i和-ii)、前b细胞生长刺激因子(pbsf)、和ip10。“c-c”趋化因子成员的例子包括但不限于单核细胞趋化蛋白1(mcp-1)、单核细胞趋化蛋白2(mcp-2)、单核细胞趋化蛋白3(mcp-3)、单核细胞趋化蛋白4(mcp-4)、巨噬细胞炎症蛋白1α(mip-1-α)、巨噬细胞炎症蛋白1β(mip-1-β)、巨噬细胞炎症蛋白1-γ(mip-1-γ)、巨噬细胞炎症蛋白3α(mip-3-α、巨噬细胞炎症蛋白3β(mip-3-β)、趋化因子(elc)、巨噬细胞炎症蛋白4(mip-4)、巨噬细胞炎症蛋白5(mip-5)、ld78β、rantes、sis-epsilon(p500)、胸腺和激活调节趋化因子(tarc)、趋化因子、i-309、人类蛋白质hcc-1/ncc-2、人类蛋白质hcc-3。
[0208]
已经鉴定了许多促进/激活内皮细胞进行血管生成的生长因子。这些包括血管内
皮生长因子(vegf a);vegf b、vegf c和vegf d;转化生长因子(tgfb);酸性和碱性成纤维细胞生长因子(afgf和bfgf);以及血小板衍生生长因子(pdgf)。vegf是一种内皮细胞特异性生长因子,具有非常特异性的作用部位,即促进内皮细胞增殖、迁移和分化。vegf是包含两个相同的23kd多肽的复合物。vegf可以作为四种不同分子量的不同多肽存在,每一种都衍生自可变剪接的mrna。bfgf是一种生长因子,可刺激成纤维细胞和内皮细胞的增殖。bfgf是单条多肽链,分子量为16.5kd。已经发现了几种分子形式的bfgf,它们的氨基末端区域的长度不同。然而,各种分子形式的生物学功能似乎是相同的。
[0209]
前体药物激活多肽也在本发明的范围内。术语前体药物激活基因是指核苷酸序列,其表达导致生成能够将非治疗性化合物转化为治疗性化合物的蛋白质,这使得细胞易于被外部因素杀死或在细胞内引起毒性状况。前体药物激活基因的一个例子是胞嘧啶脱氨酶基因。胞嘧啶脱氨酶将5-氟胞嘧啶(5fc)转化为5氟尿嘧啶(5fu),这是一种有效的抗肿瘤剂。肿瘤细胞的裂解提供了胞嘧啶脱氨酶的局部爆发,能够在肿瘤的局部点将5fc转化为5fu,从而杀死许多周围的肿瘤细胞。此外,可以使用胸苷激酶(tk)基因(参见us5,631,236和us5,601,818),其中表达tk基因产物的细胞变得易受更昔洛韦施用的选择性杀伤。前体药物激活酶的其他例子是硝基还原酶和细胞色素p450(例如cyp1a2、cyp2e1或cyp3a4)。
[0210]
治疗性抗体
[0211]
根据本发明的哑铃形载体可以包含转录盒,包括治疗性抗体或抗体片段。
[0212]
嵌合抗体是重组抗体,其中小鼠或大鼠抗体的所有v区与人抗体c区结合。人源化抗体是重组杂合抗体,其将啮齿动物抗体v区的互补决定区与人抗体v区的框架区融合。还使用了来自人抗体的c区。互补决定区(cdr)是抗体重链和轻链n端结构域内的区域,其中v区的大部分变异受到限制。这些区域在抗体分子的表面形成环。这些环提供抗体和抗原之间的结合表面。来自非人类动物的抗体会引发对外来抗体的免疫反应,并将其从环中去除。嵌合抗体和人源化抗体在施用于人类对象时都具有降低的抗原性,因为重组杂合抗体中啮齿动物(即外来)抗体的量减少,而人类抗体区域不引发免疫反应。这导致较弱的免疫反应和抗体清除率的降低。当使用治疗性抗体治疗人类疾病时,这显然是可取的。人源化抗体被设计为具有较少的“外来”抗体区域,因此被认为比嵌合抗体的免疫原性更低。
[0213]
各种抗体片段是本领域已知的。fab片段是由免疫球蛋白重链可变区和免疫球蛋白轻链可变区的免疫活性部分组成的多聚体蛋白质,它们共价偶联在一起并且能够特异性结合抗原。fab片段经由完整免疫球蛋白分子的蛋白水解切割(例如木瓜蛋白酶)生成。fab2片段包含两个连接的fab片段。当这两个片段通过免疫球蛋白铰链区连接时,会生成一个f(ab’)2片段。fv片段是由免疫球蛋白重链可变区和免疫球蛋白轻链可变区的免疫活性部分共价偶联在一起并能够特异性结合抗原的多聚体蛋白。片段还可以是仅包含一个轻链可变区的单链多肽,或其包含轻链可变区的三个cdr但没有相关的重链可变区的片段,或其包含以下三个cdr的片段重链可变区,但没有相关的轻链部分;以及由抗体片段形成的多特异性抗体,这已在例如美国专利no 6,248,516中描述。fv片段或单区域(结构域)片段通常通过在宿主细胞系中的相关鉴定区域的表达生成。这些和其他免疫球蛋白或抗体片段在本发明的范围内,并在标准免疫学教科书,例如paul,fundamental immunology(1)或janeway等,immunobiology(2)中进行了描述。分子生物学现在允许这些片段的直接合成(经由在细胞中或化学上的表达),以及它们的组合的合成。抗体或免疫球蛋白的片段也可以具有如上所
述的双特异性功能。
[0214]
rna引导的基因组编辑
[0215]
rna引导的基因组编辑基于从细菌和古生菌进化而来的rna介导的适应性防御系统,称为集群调节间隔短回文重复(crispr)/crispr相关(cas)系统,最初使用短rna来直接降解源自病毒或质粒的外来入侵dna。最常见的系统是化脓性链球菌(sp)ii型crispr系统。为了在人类细胞中编辑基因组dna,进行了几个系统调整:1.最初不同的两个短rna分子,称为crispr rna(crrna)和反式激活crrna(tracrrna),将酶引导至dna靶标以触发切割所必需的被融合形成单个引导rna(grna)。支架tracrrna结构域,以下简称cas-相互作用结构域,可以与任何crrna结构域融合,以下简称dna结合结构域(bd)。2.密码子优化将spcas9转化为hspcas9。3.为了减少脱靶编辑,引入天冬氨酸到丙氨酸取代物(d10a)以将触发hspcas9的dna双链断裂(dsb)转化为dna切口酶hspcas9n。grna的dna结合结构域(长度为20至17nt)现在可以将grna-cas9复合物引导至称为原型间隔区的互补/同源dna位点,以下称为dna靶位点,该位点必须在3’后跟随称为pam(原型间隔区相邻基序)的第二短标识符,对于此处描述的系统来说,它是5
’‑
ngg。grna的bd可以与要编辑的位点重叠,或者应该靠近该位点。然后hspcas9复合物会触发dsb,hspcas9n复合物会触发缺口。需要两个具有不同grna和移位目标位点的hspcas9n复合物来触发双切口。包括由cas9或cas9n诱导的双切口的dsb然后将激活两种内源性修复机制之一:1.在容易出错的非同源末端连接(nhej)途径中,末端将被处理和重新连接,这可能导致随机插入/删除(indel)突变。2.或者,可以提供质粒、pcr产物或单链寡脱氧核糖核苷酸(以下称为寡核苷酸)形式的修复模板,以利用触发高保真、精确编辑的同源定向修复(hdr)途径。单个刻痕使用完整链作为模板触发hdr。
[0216]
治疗性核酸
[0217]
本发明包括哑铃形载体,其表达非编码rna,包括rna适体、反式剪接rna(tsrna)和与细胞中的靶mrna序列互补的抑制性rna,以消除包括反义rna(asrna)或小或短发夹rna(shrna)或小干扰rna(sirna)或microrna(mirna)或前体mirna(pre-mirna)或反义mirna(as-mirna)的基因表达。
[0218]
适体分子折叠成允许与配体结合的结构,该配体可以是小分子、氨基酸、辅因子、肽、蛋白质、糖、脂质、核苷酸或核苷酸类似物。
[0219]
反式剪接rna分子包含三个功能性rna序列结构域:第一,靶结合结构域,其包含与一个或多个靶前体信使rna(pre-mrna)序列互补的一个或多个反义rna序列;第二,剪接结构域包含剪接供体序列或剪接受体序列、分支点和多嘧啶束;第三,编码感兴趣的肽或蛋白质的编码序列结构域。反式剪接rna分子可以特异性结合pre-mrna靶rna并启动反式剪接反应,其中反式剪接rna的编码序列结构域与靶pre-mrna连接,从而形成嵌合rna并使编码序列域的表达成为可能。
[0220]
shrna分子包含两条完全或部分互补的rna链(有义链和反义链),它们经由第三条rna连接,其中有义链和反义链彼此退火形成发夹结构的rna包含完全或部分碱基配对的茎和单链发夹环。核转录后,shrna模拟初级mirna转录物(pri-mirna)或pre-mirna,首先由drosha加工或经由exportin-5依赖性核输出途径直接从细胞核输出到细胞质中。在细胞质中,shrna由dicer加工,引导rna加载到rna诱导沉默复合物(risc)中,该复合物特异性识别和切割mrna靶标,与引导rna具有足够程度的互补性。
[0221]
sirna分子包含两条互补的rna链(有义链和反义链),它们相互退火形成双链rna分子。sirna分子通常来源于待消融基因的外显子。许多生物体通过激活导致sirna形成的级联反应来响应双链rna的存在。双链rna的存在激活了包含rnase iii的蛋白质复合物,该复合物将双链rna加工成更小的片段(sirna,长度约为21-29个核苷酸),这些片段成为核糖核蛋白复合物的一部分。sirna充当rnase复合物的引导,以切割与sirna的反义链互补的mrna,从而导致mrna的破坏。
[0222]
pre-mirna分子包含两条完全或部分互补的rna链(一个5’臂和一个3’臂,其中一个或两个都包含成熟的mirna),它们经由第三段rna连接,其中正义和反义链彼此退火以形成发夹结构的rna,其包含完全或部分碱基配对的茎和单链发夹环。核转录后,pre-mirna首先由drosha加工或经由exportin-5依赖性核输出途径直接从细胞核输出到细胞质中。在细胞质中,pre-mirna由dicer加工,成熟的mirna被加载到rna诱导沉默复合体(risc)中,该复合体特异性识别并结合与5’末端位置2至7或8互补的mrna靶标,也称为成熟mirna的种子区域,导致mrna靶标的翻译阻断或切割。
[0223]
如本文所用,术语“反义寡核苷酸”或“反义”描述一种寡核苷酸,它在生理条件下与包含特定基因的dna或与该基因的mrna转录物杂交,从而抑制该基因的转录和/或该mrna的翻译。设计反义分子以便在与靶基因杂交时干扰靶基因的转录或翻译。本领域技术人员将认识到反义寡核苷酸的确切长度及其与其靶标的互补程度将取决于所选的特定靶标,包括靶标的序列和构成该序列的特定碱基。
[0224]
优选构建和排列反义寡核苷酸以便在生理条件下选择性地与靶结合,即在生理条件下与靶序列的杂交比与靶细胞中任何其他序列的杂交更多。为了具有足够的选择性和有效的抑制作用,此类反义寡核苷酸应包含至少7个(3)个,更优选至少15个与靶标互补的连续碱基。最优选地,反义寡核苷酸包含20-30个碱基的互补序列。
[0225]
as-mirna分子是与pri-mirna靶分子、pre-mirna靶分子、成熟mirna靶或成熟mirna靶的种子区域完全或部分互补的asrna互补。优选as-mirna分子与pri-mirna或pre-mirna靶分子的dicer加工位点互补。
[0226]
dna疫苗/佐剂
[0227]
本发明包括在针对疾病和病原生物的免疫中编码抗原性多肽的哑铃形载体。通常,包含哑铃形载体的dna疫苗包括佐剂和/或载体以增强对编码抗原的免疫反应。
[0228]
几十年来,佐剂(免疫增强剂或免疫调节剂)一直用于改善对疫苗抗原的免疫反应。将佐剂掺入疫苗制剂的目的是增强、加速和延长对疫苗抗原的特异性免疫反应。佐剂的优点包括增强较弱抗原的免疫原性、减少成功免疫所需的抗原量、减少所需加强免疫的频率以及改善老年人和免疫功能低下的人对疫苗的免疫反应。选择性地,佐剂也可用于优化所需的免疫反应,例如免疫反应,例如关于免疫球蛋白类别和细胞毒性或辅助t淋巴细胞反应的诱导。此外,某些佐剂可用于促进黏膜表面的抗体反应。氢氧化铝和磷酸铝或磷酸钙已常规用于人类疫苗。最近,纳入iriv(免疫刺激性重组流感病毒体)的抗原和含有基于乳液的佐剂mf59的疫苗已在各国获得许可。佐剂可根据其来源、作用机制和物理或化学性质进行分类。最常描述的佐剂类别是凝胶型、微生物型、油乳液型和基于乳化剂的、颗粒型、合成型和细胞因子。最终疫苗产品中可能存在一种以上的佐剂。它们可以与疫苗中存在的单一抗原或所有抗原组合在一起,或者每种佐剂可以与一种特定抗原组合。目前使用或开发的
佐剂的来源和性质是高度多样化的。例如,铝基佐剂由简单的无机化合物组成,plg是一种聚合碳水化合物,病毒体可以来源于不同的病毒颗粒,mdp来源于细菌细胞壁;皂苷来源于植物,角鲨烯来源于鲨鱼肝脏,重组内源性免疫调节剂来源于重组细菌、酵母或哺乳动物细胞。
[0229]
有几种获准用于兽用疫苗的佐剂,例如对人类使用反应性太强的矿物油乳剂。类似地,完全弗氏佐剂虽然是已知的最强大的佐剂之一,但不适合人类使用。
[0230]
术语载体以下列方式解释。载体是一种免疫原性分子,当与第二种分子结合时,它会增强对后者的免疫反应。一些抗原在本质上不是免疫原性的,但当与外源蛋白质分子(如匙孔-血蓝蛋白或破伤风类毒素)结合时可能能够产生抗体反应。此类抗原含有b细胞表位,但不含t细胞表位。这种缀合物的蛋白质部分(“载体”蛋白质)提供了t细胞表位,这些表位刺激辅助t细胞,而辅助t细胞又刺激抗原特异性b细胞分化为浆细胞并产生针对抗原的抗体。
[0231]
贯穿本说明书的描述和权利要求,词语“包括”和“包含”以及词语的变体,例如“包括”和“包括”,意思是“包括但不限于”,并且不旨在(以及不)排除其他部分、添加剂、组分、整数或步骤。“本质上构成”是指具有本质整数,但包括对本质整数的功能没有实质影响的整数。
[0232]
贯穿本说明书的描述和权利要求,单数包括复数,除非上下文另有要求。特别地,在使用不定冠词的情况下,除非上下文另有要求,否则说明书将被理解为考虑复数以及单数。
[0233]
结合本发明的特定方面、实施方案或示例描述的特征、整数、特性、化合物、化学部分或基团应理解为适用于本文所述的任何其他方面、实施方案或示例,除非与之不相容。
[0234]
现在将仅通过示例并参考以下附图来描述本发明的实施例:
[0235]
图1.通用模板(ut)辅助无克隆哑铃生产的基本方案。通用dna模板是包含反向重复序列的双链dna,该反向重复序列包括表达感兴趣的编码或非编码基因所需的所有序列,例如启动子、表达盒和转录终止子。反向重复由一段核苷酸隔开。哑铃生成和纯化方案包括4个步骤。步骤1:使用5’磷酸化正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,均引入感兴趣的序列的反义或有义链;将阻断寡核苷酸b1和b2添加到反应中以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr产物稀释、热变性并缓慢冷却至室温。步骤3:哑铃结构使用单链dna连接酶共价闭合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna。虚线箭头表示转录序列。对于所有步骤:青色:( )链ut dna;洋红色:(-)负链ut dna;灰色:阻断寡核苷酸;绿色:有义(s)序列;黄色:反义(as)序列。
[0236]
图2.通用模板(ut)辅助无克隆生产shrna和mirna表达哑铃载体的方案。通用dna模板是262bp双链dna,包含99bp最小h1启动子(mh1)、聚合酶iii转录终止子(t5)和hsa-mir-30前体茎的反向重复。反向重复由四个t(正链)分隔。哑铃生成和纯化方案包括4个步骤。步骤1:使用5’磷酸化正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,均引入shrna 的反义或有义链以及shrna环序列的一半;将阻断寡核苷酸b1和b2添加到反应中以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr产物稀释、热变性并缓慢冷却至室温。步骤3:哑铃结构使用单链dna连接酶共价闭
合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna。虚线箭头表示转录序列。对于所有步骤:青色:( )链ut dna;洋红色:(-)负链ut dna;灰色:阻断寡核苷酸;绿色:shrna或mirna有义(s)序列;黄色:shrna或mirna反义(as)序列。
[0237]
图3.通用模板(ut)辅助无克隆生产较长哑铃载体的方案。通用dna模板是包含反向重复序列的双链dna,该反向重复序列包括表达感兴趣的编码或非编码基因所需的所有序列,例如启动子、表达盒和转录终止子。反向重复由一段核苷酸隔开。哑铃生成和纯化方案包括4个步骤。步骤1:使用一组正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,其中各自最外面的引物是5’磷酸化的,两组均逐步引入感兴趣的序列的反义链或有义链;将两组阻断寡核苷酸b1
n i
和b2
n i
添加到反应中以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr产物稀释、热变性并缓慢冷却至室温。步骤3:哑铃结构使用单链dna连接酶共价闭合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna。虚线箭头表示转录序列。对于所有步骤:青色:( )链ut dna;洋红色:(-)负链ut dna;灰色:阻断寡核苷酸;绿色:有义(s)序列;黄色:反义(as)序列。
[0238]
图4.通用模板(ut)辅助无克隆生产非编码rna表达哑铃载体的方案。通用dna模板是包含反向重复序列的双链dna,该反向重复序列包括表达感兴趣的非编码基因所需的所有序列,例如启动子、表达盒和转录终止子。反向重复由一段核苷酸隔开。哑铃生成和纯化方案包括4个步骤。步骤1:使用一组正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,其中各个最外面的引物是5’磷酸化的,如果要表达非回文序列,则可以引入转录终止子的反向互补序列,两组均逐步引入感兴趣的序列的反义链或有义链;将两组阻断寡核苷酸添加到反应中以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr产物稀释、热变性并缓慢冷却至室温。步骤3:哑铃结构使用单链dna连接酶共价闭合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna。虚线箭头表示转录序列。对于所有步骤:青色:( )链ut dna;洋红色:(-)负链ut dna;灰色:阻断寡核苷酸;绿色:有义(s)序列;黄色:反义(as)序列。
[0239]
图5.通用模板(ut)辅助无克隆生产编码rna表达哑铃载体的方案。通用dna模板是包含反向重复序列的双链dna,该反向重复序列包括表达感兴趣的编码基因所需的所有序列,例如启动子、表达盒和转录终止子。反向重复由一段核苷酸隔开。哑铃生成和纯化方案包括4个步骤。步骤1:使用一组正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,其中各个最里面的引物引入翻译起始密码子(5’cat3’)的反向互补序列,相应的最外面的引物是5’磷酸化的,引入了翻译终止密码子的反向互补序列,两套引物都逐步引入感兴趣的序列的反义链或有义链;将两组阻断寡核苷酸b1
n i
和b2
n i
添加到反应中,以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr产物稀释、热变性并缓慢冷却至室温。步骤3:哑铃结构使用单链dna连接酶共价闭合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna。虚线箭头表示转录序列。对于所有步骤:青色:( )链ut dna;洋红色:(-)负链ut dna;灰色:阻断寡核苷酸;绿色:有义(s)序列;黄色:反义(as)序列。
[0240]
图6.通用模板(ut)辅助无克隆生成哑铃形双表达载体的基本方案。通用dna模板
是一种双链dna,它包含两个启动子,每条链一个启动子,它们可以相同或不同,还可能包含其他调节序列。哑铃生成和纯化方案包括4个步骤。步骤1:使用5’磷酸化正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,均引入感兴趣的序列(相同或不同的序列)、转录终止子的反向互补序列(相同或不同)、一个或多个脱碱基位置(空位)和能够形成发夹结构的5’末端序列;将阻断寡核苷酸b1和b2添加到反应中以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr反应冷却至室温。步骤3:哑铃结构使用双链dna连接酶共价闭合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna。虚线箭头表示转录序列。对于所有步骤:青色:ut dna;黄色和绿色:感兴趣的序列;洋红色:无碱基引物位点(空位);灰色:阻断寡核苷酸。
[0241]
图7.通用模板(ut)辅助无克隆生成哑铃形双表达载体的详细方案。通用dna模板是包含两个启动子p1和p2的双链dna,每条链中一个启动子,它们相同或不同。哑铃生成和纯化方案包括4个步骤。步骤1:使用5’磷酸化正向(fw)和/或反向(rv)引物对通用模板进行pcr扩增,均引入感兴趣的序列soi1和soi2(相同或不同序列),转录终止子的反向互补序列t1(-)(相同或不同),一个或多个脱碱基位置(空位),以及一个能够形成发夹结构的5’末端序列;将阻断寡核苷酸b1和b2添加到反应中以抑制模板重折叠并促进引物结合。虚线表示ut之外的质粒序列。步骤2:对于哑铃结构预折叠,将pcr反应冷却至室温。步骤3:哑铃结构使用双链dna连接酶共价闭合。步骤4:用t7 dna聚合酶处理去除寡核苷酸和未连接的哑铃dna,产生共价闭合的哑铃载体dna,包含两个表达盒p1( )-soi1-t1( )和p2( )-soi2-t2( ),用于表达感兴趣的soi1和soi2序列。虚线箭头表示转录序列。对于所有步骤:青色:ut dna;黄色和绿色:感兴趣的序列;洋红色:无碱基引物位点(空位);灰色:阻断寡核苷酸。
[0242]
图8.通用模板(ut)的序列、克隆和验证。(a),ut加链序列。青色:mir-30 5’和3’茎;洋红色:转录终止子(有义和反义);橙色:成环四核苷酸序列;黑色:mh1序列(有义和反义)。(b),ut克隆方案。5’磷酸化寡核苷酸odn2和odn3分别与寡核苷酸odn1和odn4退火。将得到的双链体连接起来,并使用hindiii和bamhi限制性位点将双链ut序列克隆到pvax1中。青色:ut正链;洋红色:ut负链。(c),分析性1%琼脂糖凝胶电泳示出退火的寡核苷酸对odn1/2和odn3/4以及连接的杂合体。(d)克隆质粒pvax1-ut的分析hindiii和/或bamhi消融和1%琼脂糖凝胶电泳。hindiii/bamhi双酶切指示262bp ut条带。(e),pvax1-ut的hindiii和bamhi克隆位点的测序。
[0243]
图9.基于pcr的通用模板(ut)辅助哑铃生产。(a)ut的pcr扩增取决于阻断寡核苷酸的添加。在不添加阻断寡核苷酸(-)的情况下,未观察到pcr扩增,仅检测到126bp重折叠的ut单链。当添加阻断寡核苷酸( )时,pcr扩增的双链哑铃dna(303bp)和重折叠的哑铃单链(146bp)均被检测到。(b)pcr条件的优化。pcr产率实际上与52℃至65℃范围内的退火温度无关。添加5%dmso提高了双链哑铃dna的产量。(c)参考图1中定义的步骤1至4评估dna产品。步骤1:pcr ut扩增产生双链哑铃dna(303bp)和重新折叠的哑铃单链(146bp);步骤2:热变性和重折叠将双链哑铃dna转化为哑铃单链;步骤3:如果5’磷酸化引物用于pcr,单链dna连接共价闭合哑铃载体dna;步骤4:核酸外切酶处理去除未连接的dna并产生共价闭合的哑铃载体(146bp)。
[0244]
图10.完整的正链和负链衍生的荧光素酶靶向哑铃载体的序列和结构。核纤层蛋白a/c靶向载体具有相同的哑铃核心。
[0245]
图11.哑铃载体生成。通过pcr产物的5’磷酸化(左侧)或使用用于pcr反应的5’磷酸化引物(右侧)生成146bp哑铃载体dna。步骤是指图1中定义的步骤1至4。
[0246]
图12.使用细胞内facs监测的正( )和负(-)链衍生的核纤层蛋白靶向(lam)哑铃(db)载体对hek 293t细胞中核纤层蛋白a/c的击倒。(a)哑铃载体和转录核纤层蛋白a/c靶向shrna的序列和结构。根据mfold和rnafold的预测绘制shrna二级结构。(b-d)一项实验的代表性直方图叠加。(b)染色(初级抗核纤层蛋白a c抗体加二级驴抗兔igg h&l)与未染色(仅初级抗核纤层蛋白a c抗体)未转染的活细胞。(c)由0.1、0.5或2.5pmol加上链衍生的抗核纤层蛋白a/c shrna表达db载体[lam( )db]或3pmol抗核纤层蛋白a/c阳性对照sirna(lam-sirna)触发的击倒。(d)由表达db载体[lam(-)db]或3pmol lam-sirna的0.1、0.5或2.5pmol减去链衍生的抗核纤层蛋白a/c shrna触发的击倒。(e-g)染色hek293t细胞中核纤层蛋白a/c相对于由核纤层蛋白a/c染色细胞(e)的分数表示的未转染细胞(100%)的击倒,核纤层蛋白a/c染色细胞(f)的几何平均荧光强度(gmfi)和核纤层蛋白a/c染色细胞(g)的中值荧光强度。对照哑铃(对照db)是1:1混合的正链和负链衍生荧光素酶靶向哑铃dna。值是三个独立实验的平均值
±
sem。使用学生t检验进行统计分析。p值表示相对于染色无转染对照的显着性。(b-g)ntc:无转染对照;无dna对照:缓冲待转染细胞。
[0247]
图13.正(a)和负(b)链衍生荧光素酶靶向哑铃的转化率测定。使用非磷酸化正向引物和5
’‑
磷酸化反向引物进行pcr。使用100u环连接酶在50^l反应体积中对6^g pcr产物进行变性、重折叠和连接。用核酸外切酶处理连接的重折叠哑铃dna,并使用1%琼脂糖凝胶电泳分析核酸外切酶前后直接可比较的样本。只有成功连接的共价闭合哑铃dna才能抵抗核酸外切酶处理。通过使用imagej v1.48软件量化条带强度来确定将未连接到连接的整体哑铃dna的转化率。由于理论上每个反应中只有正链或负链来源的哑铃dna,即总dna的50%可以连接,因此正链或负链来源的哑铃dna的实际转化率是检测到的总转化率。
[0248]
图14.通过正( )和负(-)链衍生的荧光素酶靶向(luc)哑铃(db)载体击倒hek293t细胞中的萤火虫荧光素酶。(a)哑铃载体和转录荧光素酶靶向shrna的序列和结构。根据mfold和rnafold的预测绘制shrna二级结构。(b)使用磷酸化正向(5’p-fw)或反向(5’p-rv)引物选择性生成( )或(-)链衍生db载体。步骤参考图1中定义的步骤1至4。(c、d)正( )和/或负(-)链衍生荧光素酶靶向db载体的功能验证。用萤火虫荧光素酶报告载体pgl3和0.5或1.5pmol哑铃载体dna共转染细胞。ntc:无转染对照。萤火虫荧光素酶mrna(c)或表达(d)水平相对于未抑制的阴性对照在转染后48小时使用rt-qpcr或荧光素酶报告基因测定进行测量。相对rna水平根据折叠变化(2-δδct
)计算,其中δct=ct luciferase
–
ct
β-actin
。值是三个独立实验的平均值
±
sem。使用学生t检验(c)或重复单向方差分析和tukey事后多重比较检验检测(d)进行统计分析。
[0249]
图15.核酸外切酶处理后哑铃载体的纯度对比。(a),正( )和负(-)链衍生荧光素酶(luc)和核纤层蛋白a/c(lam)靶向哑铃(db)载体的1%琼脂糖凝胶电泳。(b),lam(-)db dna的毛细管凝胶电泳(生物分析仪,安捷伦)。鉴定的136bp峰指的是146bp哑铃载体dna。检测到的和预期的载体大小的差异是指哑铃载体dna的凝胶延迟与dna梯状标记的双链dna片段的凝胶延迟略有不同。
[0250]
图16.通过细胞内荧光激活细胞分选(facs)监测核纤层蛋白a/c表达。非转染细胞(ntc)中的细胞核纤层蛋白a/c表达用抗核纤层蛋白a c主要抗体标记(左图:未染色),然后
用驴抗兔igg h&l alexa fluor 647-a二级抗体染色(右图:染色)。活hek293t细胞(a)、alexa fluor 647-a散点图(b)和直方图(c)的门控。
[0251]
序列表
[0252]
seq id no 1:
[0253][0254]
seq id no 2:
[0255][0256]
seq id no 3:
[0257][0258]
seq id no 4:
[0259][0260]
seq id no 5:
[0261][0262]
seq id no 6:
[0263][0264]
seq id no 7:
[0265][0266]
seq id no 8:
[0267]
[0268]
seq id no 9:
[0269][0270]
seq id no 10:
[0271][0272]
seq id no 11:
[0273][0274]
seq id no 12:
[0275][0276]
seq id no 13:
[0277][0278]
seq id no 14:
[0279][0280]
seq id no 15:
[0281][0282]
seq id no 16:
[0283][0284]
seq id no 17:
[0285][0286]
seq id no 18:
[0287][0288]
seq id no 19:
[0289][0290]
seq id no 20:
[0291][0292]
seq id no 21:
[0293][0294]
seq id no 22:
[0295][0296]
seq id no 23:
[0297][0298]
seq id no 24:
[0299][0300]
seq id no 25:
[0301][0302]
材料和方法
[0303]
寡脱氧核糖核苷酸(odn)和引物
[0304]
通用模板。由于内部的自我互补性,通用模板不能通过基因合成产生,而是由两对互补的寡脱氧核糖核苷酸(idt,skokie,il)组装而成(图s1a,b):
[0305][0306]
粗体:hindiii和bamhi兼容的悬垂;下划线:哑铃环形成四核苷酸。5’磷酸化寡核苷酸ut2和ut3分别与互补寡核苷酸ut1和ut4杂交。将得到的ut1/ut2和ut3/ut4双链体连接形成带有hindiii和bamhi兼容的5
’‑
突出端的通用模板序列,随后将其克隆到pvax1(thermo fisher scientific,waltham,ma)中,产生通用模板载体pvax1-ut(图s1b)。对于克隆,我们使用reca缺陷型大肠杆菌菌株top10。通用模板的克隆通过pcr和fastdigest bamhi/hindiii(thermo fisher scientific,waltham,ma)核酸内切酶切割,随后通过分析琼脂糖凝胶电泳得到预期的262bp插入大小和连接位点的测序得到证实(图s1c,d)。由于高度的自我互补性,完整通用模板的测序不成功。
[0307]
用于生产萤火虫荧光素酶或核纤层蛋白a/c靶向shrna表达哑铃的引物。萤光素酶或核纤层蛋白a/c特异性引物由aitbiotech(新加坡)或idt(新加坡)合成。大写字母表示通用模板结合位点,小写字母表示shrna编码序列,其中环形成核苷酸加下划线:正向引物:fp_luciferase 5
’‑
tgaaggctcctcagaaacagctccgcgctcactgagaagattt-3’;5
’‑
fp_lamin 5
’‑
tgaaagcccagatcgtcaccacccgccgcgctcactgagaagattt-3’.。反向引物:rp_luciferase 5’agagaggctcctcagaaacagctctttgcctactgagaagatttttctgt-3’,;rp_lamin5’agagaagcccagatcgtcaccacctttttgcctactgagaagatttttctgt-3’.。
[0308]
阻断odn:将两个阻断odn(idt,skokie,il)添加到pcr以抑制通用模板链的重折叠和自引发。
[0309]
阻断_1:
[0310][0311]
阻断_2:
[0312][0313]
定量逆转录pcr(rt-qpcr)的引物。用于荧光素酶和β-肌动蛋白mrna水平定量的引物由aitbiotech(新加坡)合成。pcr正向引物:qpcr_fp_luciferase 5
’‑
cgctgggcgttaatcaaaga-3’;qpcr_rp_β-actm 5
’‑
ctggcacccagcacaatg-3’.。逆转录和pcr反向引物:qpcr_rp_luciferase 5
’‑
gtgttcgtcttcgtcccagt-3’;qpcr_rp_β-actin 5
’‑
gccgatccacacggagtact-3’.。
[0314]
引物磷酸化。对于链特异性哑铃载体的生成,正向或反向引物都是5
’‑
磷酸化的。每个50pmol引物与10u t4多核苷酸激酶(thermo fisher scientific,waltham,ma)在1mm atp存在下在37℃下孵育20分钟,然后在75℃下热灭活酶10分钟。
[0315]
哑铃载体生成
[0316]
哑铃载体dna的pcr扩增。使用1u taq dna聚合酶(invitrogen)、1.0μm的每种引物和阻断odn、0.2mm的每种dntp(invitrogen)、100ng hindiii/bamhi裂解的pvax1-ut,5%v/v dmso(thermo fisher scientific,waltham,ma),反应体积为30-50μl,在1x taq dna聚合酶缓冲液(invitrogen)中对通用模板和shrna编码dna的附件进行pcr扩增。pvax1-ut的线性化通常会提高pcr产量,但不是必需的。热循环如下进行:在96℃初始变性5分钟;变性(95℃,30秒)、退火(59℃,30秒)和延伸(72℃,1分钟)的27次循环;最后在72℃延伸10分钟。50ml pcr反应产生约10μg dna。
[0317]
链分离和退火。pcr产物通过基于硅胶膜的离心柱(qiaquick pcr纯化试剂盒,qiagen,hilden,germany)纯化。将纯化产物在1x杂交缓冲液(1m nacl、100mm mgcl2和
200mm tris-hcl,ph 7.4)中稀释至400μl,在96℃热变性5分钟,然后逐渐冷却至室温以允许用于正链和/或负链哑铃载体的分子内折叠。使用乙醇沉淀浓缩所得dna,通过离心沉淀,并重新悬浮在无核酸酶水中。
[0318]
单链环dna的连接。将1至6μg(~10至60pmol)的dna与2.5mm mncl2、1m甜菜碱(sigma,st.louis,mo)和50至100u环连接酶ii(epicenter,madison,wi)在60℃的1x环连接酶ii反应缓冲液中16小时,然后在80℃加热灭活连接酶10分钟。当用100u环连接酶连接6μg dna时,观察到最高的转化率。
[0319]
核酸外切酶处理。连接后,用10u的t7 dna聚合酶(thermo fisher scientific,waltham,ma)在37℃下处理1小时,然后在80℃下热灭活10分钟。产品在10%天然聚丙烯酰胺凝胶或1%琼脂糖凝胶上进行评估,电泳后用溴化乙锭染色和/或使用苯酚-氯仿-异戊醇(25:24:1)提取(1x)、氯仿-异戊醇(24:1)进行纯化再萃取(3x)和乙醇沉淀。
[0320]
靶基因敲低检测
[0321]
荧光素酶击倒测定。hek293t细胞维持在dulbecco改良eagle培养基(hyclone,south logan,ut)中,添加10%(v/v)胎牛血清(hyclone,south logan,ut)和1%青霉素-链霉素抗生素溶液(thermo fisher scientific,waltham,ma)。转染前24小时,将2x104个细胞/孔接种在96孔板中。使用lipofectamin 2000(thermo fisher scientific,waltham,ma)和1.5pmol或0.5pmol的正链或负链哑铃载体dna将细胞与100ng荧光素酶表达质粒pgl3(promega,madison,wi)共转染,试剂:dna比例为1:2.5。对于阳性对照(仅pgl3),使用空pvax1(thermo fisher scientific,waltham,ma)作为饲养层dna,以确保所有细胞接受相同数量的dna。转染后48小时,用无菌pbs洗涤细胞并在20μl被动裂解缓冲液(promega,madison,wi)20分钟,轻轻摇动。用50μl larii试剂(promega,madison,wi)处理10μl裂解液,并在biotek读数器(biotek instruments,winooski,vt)上对荧光进行量化。
[0322]
通过细胞内荧光激活细胞分选(facs)监测核纤层蛋白a/c击倒。如上所述,在转染前24小时将hek293t细胞培养并接种在96孔板中。根据制造商的方案,使用lipofectamine3000(thermo fisher scientific,waltham,ma)用0.1、0.5或2.5pmol哑铃载体dna或3pmol sigenomelamin a/c对照sirna(dharmacon,lafayette,co)转染细胞。转染后24小时更换培养基,48小时后收获细胞。对于facs分析,吸出培养基并用pbs冲洗细胞一次,然后用50μl的1x胰蛋白酶-edta(gibco)进行胰蛋白酶消化。通过在200μl培养基中以4200rpm离心6分钟收集胰蛋白酶处理的细胞。在细胞内染色之前,根据制造商的方案,将沉淀的细胞重新悬浮在100μl培养基中,用细胞内固定和透化缓冲液组(ebioscience,san diego,ca)固定和透化。为了评估核纤层蛋白a/c击倒,细胞核纤层蛋白a/c被抗核纤层蛋白a c抗体(ab133256)(1/200)和驴抗兔igg h&l af647(ab150075)(1/200)(abcam,cambridge,uk)染色。facs在lsrfortessa细胞分析仪上进行,facsdiva软件v6.1.3(bd biosciences,san jose,ca)用于采集样本。flowjo软件v10.5.2(tree star,ashland,or)用于数据分析。
[0323]
计算二级结构预测
[0324]
使用算法mfold和/或rnafold
17,18
折叠dna和rna的最小自由能二级结构。
[0325]
统计分析
[0326]
图表代表三个独立实验的平均值
±
sem。使用重复单向方差分析和tukey的事后多
重比较测试(荧光素酶击倒数据)或使用学生t检验(核纤层蛋白a/c击倒数据)进行统计分析。graphpad prism版本7软件(graphpad,la jolla,ca)用于统计分析。p值如所示。
实施例:
[0327]
用于生成哑铃形载体的通用模板辅助、无克隆和无核酸内切酶方法
[0328]
在当前最先进的方案中,每个新哑铃载体的生成都始于将要实施到哑铃中的感兴趣序列单独克隆到质粒载体中。本发明的方法只需要制备一种通用模板,包括启动子、增强子、dna核定位信号、内含子、转录终止子、rna核输出信号、wpre等调控序列。然后经由化学合成的pcr引物引入感兴趣的序列,无需进一步克隆,也不需要核酸内切酶(图1-7)。这种生成的哑铃可以用作分子诱饵或表达载体。表达载体可以表达非编码rna,包括shrna、pre-mirna、mirna、适配体、反义rna和反义mirna(图1-4、6、7)和/或表达肽或蛋白质编码rna(图5-7)。发夹结构rna(如shrna和pre-mirna)的表达盒可以设计为发夹模板转录表达盒,其中表达盒类似于转录rna的发夹结构。在这些载体中,冗余序列被消除,转录围绕哑铃环之一进行。可以使用本发明的方法生成发夹模板转录哑铃载体(图1-5)。
[0329]
新方法基于通用dna模板的pcr扩增,对于shrna表达,该模板包含(i)最小h1启动子
15
、(ii)聚合酶iii转录终止子(t5)以及(iii)hsa-mir-30前体茎(图8a)。据报道,hsa-mir-30茎可促进shrna加工,并已在哑铃载体设计中成功实施
13,16
。一旦生成,通用模板可用于无克隆生成任何表达shrna的哑铃载体。在pcr期间通过pcr引物引入编码相应小rna表达的序列(步骤1)。无论pcr引物的小rna特异性5’部分如何,它们都具有相同的3’末端靶结合位点,这有助于并行pcr扩增。通用dna模板的两条链都具有高度的自我互补性,为了改善其扩增,将封闭寡核苷酸添加到pcr反应中以抑制变性dna的分子内重折叠并支持引物结合。得到的双链pcr产物的两条dna链( 和-)中的每一条在稀释、热变性和分子内重折叠后都会产生一个5’和3’末端悬空的开放哑铃支架(步骤2)。然后使用单链dna(ssdna)连接酶连接这些末端(步骤3)。所有带有5’或3’末端的dna分子都通过核酸外切酶消化去除,产生干净的共价闭合哑铃载体(步骤4)。决定使用一个或两个5
’‑
磷酸化pcr引物后,正链、负链或两条链都会产生哑铃载体。
[0330]
由于高度的自我互补性,通用模板的生成具有挑战性,所有通过基因合成生成通用模板的尝试都失败了。相反,通用模板由两对互补的寡脱氧核糖核苷酸(oligos)组装而成,其中自互补序列部分彼此分离(图8a-c)。对互补的寡核苷酸对进行退火,每个形成一个互补的3
’‑
悬垂和hindiii或bamhi 5
’‑
悬垂。然后首先使用粘合剂3’末端连接成对的退火寡核苷酸,凝胶纯化,并使用hindiii和bamhi克隆位点插入克隆载体pvax1,从而产生通用模板载体pvax1-ut。通过分析限制性内切酶切割和随后的片段凝胶电泳以及测序证明了通用模板的成功克隆:hindiii/bamhi双消化产生了预期的262bp插入大小;由于插入物的自我互补性,完整插入物的测序不成功,但可以对克隆位点进行测序(图8d,e)。
[0331]
接下来,我们的目标是使用引物对通用模板进行pcr扩增,这些引物引入了针对已发表的萤火虫荧光素酶靶向shrna的序列编码9。然而,通用模板的内在自我互补性阻碍了常规pcr扩增,而传统pcr扩增并未产生任何预期的大小。在pcr反应中加入两个长封闭寡核苷酸后观察到产物(图9a)。这些封闭寡核苷酸的设计使它们与通用模板的正链或负链的相应5’半互补,从而抑制分子内链重折叠并促进引物与模板dna的3’末端结合(图2)。因为阻
断寡核苷酸与通用模板序列结合,它们代表了这个哑铃生成方案中一个恒定的、目标和shrna独立的组件。获得的pcr产物与双链通用模板(303bp)和重折叠单链(146bp)大小相对应。在pcr反应中添加5%(v/v)dmso会产生更多更大的产物,这表明扩增更有效,因为引物结合和延伸与单链重折叠更成功地竞争(图9b)。然后对纯化的pcr产物进行热变性和再折叠,产生更多的发夹结构单链(图9c,泳道2)。正如预期的那样,ssdna结扎(泳道3)和随后的核酸外切酶消化(泳道4)仅在5
’‑
磷酸化引物用于pcr时才产生抗核酸外切酶的哑铃载体dna(图9c)。
[0332]
在决定使用5
’‑
磷酸化正向引物、5
’‑
磷酸化反向引物或两种磷酸化引物进行pcr时,仅(i)正链衍生哑铃,或(ii)负链衍生哑铃,或(iii)可以生成两者的混合(图2和10)。为了获得正链和负链衍生哑铃的混合物,如果pcr引物或pcr产物的5’末端被磷酸化,则没有区别(图11)。在这个例子中,正负链衍生的哑铃和表达的shrna非常相似,但并不相同,因为它们在环和hsa-mir-30茎中的序列和结构不同(图12a和10)。mirna茎区的不对称性归因于这样一个事实,即只有当发夹模板转录哑铃也具有相应的错配时,才能实现部分错配的mirna前体rna的正确转录。因此,只有正链衍生的哑铃表达了用原始mir-30茎延伸的shrna(图12a)。由负链衍生的哑铃表达的shrna延伸有由mir-30的反义序列形成的mir样茎,并带有一个环,该环代表正链衍生的shrna中环的反向互补。观察到的转化率,即重新折叠的146bp哑铃载体dna成功连接并抵抗随后的核酸外切酶处理的部分,对于正链或负链衍生的荧光素酶靶向哑铃的生产,其被测量为34%或28%(图13)。考虑到只有一种pcr引物被磷酸化来产生这些哑铃,因此理论上只有一半的重折叠dna可以被连接,那么可连接的正链或负链衍生的哑铃dna的实际转化率为68%或56%。在这些反应中,我们使用100u的环连接酶连接了6μg dna。
[0333]
使用上述方案,使用磷酸化的正向或反向引物,我们在单独的反应中生成了正负链衍生的荧光素酶或核纤层蛋白a/c靶向哑铃(图12和14)。使用琼脂糖凝胶电泳控制核酸外切酶处理后载体的纯度(图15a)。额外的毛细管凝胶电泳确定负链衍生的核纤层蛋白a/c靶向哑铃的纯度为83%(图15b)。为了测量哑铃载体触发的荧光素酶击倒,使用lipofectamine2000将hek293t细胞与荧光素酶表达载体pgl3-对照和0.5或1.5pmol正链或负链衍生的哑铃载体dna共转染。转染后48小时,萤火虫荧光素酶mrna和活性水平相对于pgl3-对照载体进行量化(图12c,d)。两个哑铃都触发了显着的剂量依赖性荧光素酶击倒,这在负链衍生的哑铃载体的情况下更为明显,表明非天然mir样茎是功能性的。在1.5或0.5pmol载体dna时,由正链衍生的哑铃触发的击倒为85%(p《0.001)或50%(p《0.001),在1.5或0.5pmol dna时,负链衍生的哑铃触发的的击倒为97%(p《0.001)或相对于pgl3阳性对照为75%(p《0.001)。为了研究核纤层蛋白a/c的击倒,hek293t细胞用0.1、0.5或2.5pmol正链或负链衍生的哑铃载体dna转染,或用3pmol sigenome核纤层蛋白a/c阳性对照sirna或0.5pmol荧光素酶靶向哑铃对照载体dna(正链和负链衍生哑铃的1:1混合物)使用lipofectamine 3000转染。转染后48小时,使用兔抗核纤层蛋白a c一抗和驴抗兔对细胞内核纤层蛋白a/c进行染色,通过流式细胞术分析监测igg h&l af647二抗和核纤层蛋白a/c击倒(图14,16)。虽然正链衍生的哑铃在2.5或0.5pmol dna时触发了显着的、剂量依赖性的核纤层蛋白a/c击倒,但用负链衍生的哑铃观察到的击倒不太明显。
[0334]
此处描述的方案结合了先前报告的哑铃载体生产方案的所有优点。它代表(i)无
克隆方案,(ii)不涉及任何限制性或切口内切核酸酶,(iii)采用有效的分子内连接反应,以及(iv)允许产生极小的发夹模板转录哑铃载体。先前描述的空位引物pcr方案也涉及分子内连接,但需要一个克隆步骤来生成每个新载体,并且由于无碱基序列位置的存在,它不适合生成发夹模板转录载体
14
。相反,jiang等描述的方法,适用于生产发夹模板转录哑铃,但需要限制性和切口内切核酸酶,并且涉及效率较低的分子间连接反应
9,13
。用于此处报告的方案的pcr引物也始终具有相同的3’末端模板结合位点作为一个5’末端序列,它依赖于相应的小rna并随其变化,但在每个相应的引物对中在很大程度上是相同的。因此,引物退火温度始终相同,并且可以广泛排除引物二聚体的形成,这两者都有助于使用单个循环程序进行并行pcr反应。随后的连接反应代表一种分子内连接,与涉及分子间环连接的替代方案相比,它通常更有效。作为推论,该方法观察到的转化率高于使用分子间连接反应的方案所报告的转化率。对于空位引物pcr方法,使用t4 dna连接酶连接双链带缺口哑铃dna时,转化率高达92%平;然而,当使用环连接酶将悬垂的单链5’末端与碱基配对的3’末端连接时,使用空位引物pcr方法仅观察到略高的75%的转化率。使用此处描述的方法生产的哑铃dna的纯度在使用空位引物pcr方法生产的载体的82%至94%的纯度范围内。未来的临床前和临床应用将需要额外的纯化步骤。
[0335]
我们证明了这种新方法可以生成部分不匹配的表达shrna的哑铃载体的原理证明,表明该技术也可能被探索用于生成表达mirna的哑铃。较早报道了哑铃载体中的错配并证明不会损害载体活性
13
。相反,发现末端单核苷酸错配可改善核靶向和哑铃形表达载体的活性
14
。
[0336]
我们观察到,在荧光素酶或核纤层蛋白a/c靶向哑铃中,负链或正链衍生的哑铃分别表现出更强的靶基因敲低活性。这种差异可能归因于dicer处理内源性shrna的效率和准确性方面的差异,这取决于shrna环和茎的序列和结构。在这里,我们使用了hsa-mir-30茎,因为据报道mirna茎在大多数情况下促进shrna加工和击倒活动
16
。因此,尽管相应的正链和负链衍生的哑铃编码相同的引导序列,转录的如上所述,小发夹rna包含不同的microrna茎和不同的环。因此,dicer加工的差异可能导致不同的指导rna水平和/或指导rna序列的确切5’和3’终止子的差异。这些差异可以解释这样的观察结果,即根据靶序列和相应的指导rna序列和结构,正链或负链衍生的哑铃会触发更强的靶基因击倒。然而,当放弃包含mirna茎并同时考虑回文环序列时,正链和负链衍生的哑铃将是相同的,并且单个反应将仅生成单个载体。
[0337]
总之,这种新方法可以在短时间内以低成本有效地生成尺寸最小的发夹模板转录哑铃,并且可以探索用于功能基因组筛选或药物开发的shrna或mirna表达载体的并行生产。
[0338]
参考文献
[0339]
1.biasco,l.,baricordi,c.and aiuti,a.(2012).retroviral integrations in gene therapy trials.mol.ther.20,709-716.
[0340]
2.yin,h.,kanasty,r.l.,eltoukhy,a.a.,vegas,a.j.,dorkin,j.r.and anderson,d.g.(2014).non-viral vectors for gene-based therapy.nat.rev.genet.15,541-555.
[0341]
3.mok,p.l.,cheong,s.k.,leong,c.f.,chua,k.h.and ainoon,o.(2012)
h1rna gene.nucleic acids res.29,2502-2509.
[0354]
16.zeng,y.,wagner,e.j.and cullen,b.r.(2002).both natural and designed micro rnas can inhibit the expression of cognate mrnas when expressed in human cells.mol.cell.9,1327-1333.
[0355]
17.zuker,m.(2003).mfold web server for nucleic acid folding and hybridization prediction.nucleic acids res.31,3406-3415.
[0356]
18.hofacker,i.l.(2003).vienna rna secondary structure server.nucleic acids res.13,3429-3431.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。