1.本发明属于化学合成领域,具体而言,涉及精细化工领域中胺化反应以及胺化反应中的催化剂,更具体而言,涉及使用环内酯的胺化反应及其使用的催化剂。
背景技术:
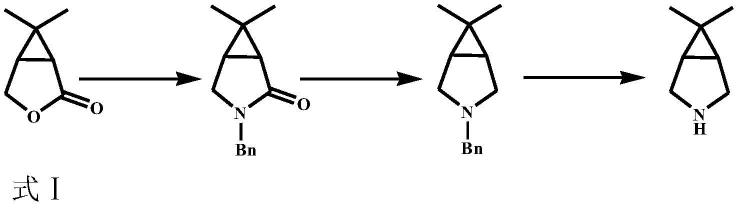
2.通过式ii所示的双环内酰胺化合物可以用来制备6,6-二甲基-3-氮杂双环[3.1.0]己烷,已知的是,6,6-二甲基-3-氮杂双环[3.1.0]己烷(6,6-dimethyl-3-azabicyclo[3.1.0]hexane;cas号:943516-54-9)是一种重要的医药中间体,它是很多药物如丙肝蛋白酶抑制剂博赛泼维(boceprevir)和治疗新冠病毒的口服药(pf-07321332)合成过程中所使用的重要原料。
[0003][0004]
作为式ⅱ双环内酰胺化合物的制备方法,已经公开的方法包括:
[0005]
住友化学株式会社公开的引用文献1的实施例1中公开了式ⅰ化合物在水的存在下(相对于式ⅰ化合物1摩尔,水的使用量为30摩尔),使用苄胺在温度为180℃下,压力为0.7mpa的条件下反应10小时,并经后处理得到苄基取代的双环内酰胺化合物(收率为94.3%)。该苄基取代的双环内酰胺化合物经氢化锂铝还原酰胺后还需进一步加氢脱苄后才能得到6,6-二甲基-3-氮杂双环[3.1.0]己烷化合物。具体过程参见如下反应过程:
[0006][0007]
其中,如果式i化合物直接用nh3作为胺源,则胺化试剂较苄胺的原料成本大大下降,同时也省去了脱苄步骤,原子经济性更高,使6,6-二甲基-3-氮杂双环[3.1.0]己烷的合成效率大大提高,例如下述合成路线。
[0008][0009]
目前通过催化剂使内酯进行胺化反应的研究被大量报道,催化剂的使用可以使胺化反应在相对温和的条件下进行,同时使反应时间缩短反应压力降低。
[0010]
例如,例如引用文献2介绍了采用固定床反应器,以so
42-/mxoy型固体超强酸催化剂,1,4丁内酯和苯胺反应合成n-苯基吡咯烷酮,最佳条件为:原料配比为1.2:1、反应温度为300℃、进料速度为1.2ml/min。产物经重结晶分离、抽滤、烘干,得到白色晶体,npp的转化率为98.7%。
[0011]
引用文献3公开了以内酯为原料,与一级脂肪胺或者液氨进行氨化置换,反应生成相应的含c=o键的含单个氮杂环化合物,过程中使用催化剂为多相催化剂,所述多相催化剂为so
42-/mxoy型超强酸、阳离子树脂、氧化铌中的一种,所述的mxoy选自al2o3、tio2、zro2、fe3o4中的一种或者一种以上;反应条件为150~270℃,0.5~8.0mpa,脂肪胺或者氨与内酯的摩尔比例为1~3,反应在釜式或管式反应器中进行。
[0012]
引用文献4公开了包括在(a)内酯、(b)胺与/或氨、及(c)水存在下,利用晶态铝硅酸盐沸石作为催化剂,在气相进行胺化反应,所用内酯及胺与/或氨的摩尔比通常为1∶0.5至1∶30,所用内酯及水的摩尔比通常为1∶0.5至1∶20,温度为180至400℃,反应压力为0atm至10atm。
[0013]
引用文献5公开了一种n-羟乙基吡咯烷酮(nhp)的气相制备方法,以碱金属、碱土金属硝酸盐对y型分子筛进行浸渍改性,得到不同金属阳离子组成的y型分子筛;并以改性y-型分子筛作为催化剂,在240℃~300℃、气相、常压条件下实施γ-丁内酯与乙醇胺反应制备n-羟乙基吡咯烷酮(nhp),产物单程收率达到46%以上,总收率可达到90%以上。
[0014]
引用文献6中介绍了用zsm分子筛催化剂制备n-甲基吡咯烷酮(nmp),采用固定床反应装置,原料甲胺水溶液和γ-丁内酯(gbl)按比例混合后,以液相形式进入反应器。反应器上下段均填充一定量的碳化硅颗粒,下段填料足够长,作为预热段,以确保反应在气相状态下进行,其合成n-甲基吡咯烷酮的最佳条件为:采用硅铝摩尔比为120的zsm-5分子筛为催化剂,反应温度300℃、n(甲胺):n(gbl):n(水)=1.5:1:30、常压、空速0.5h-1
、产率99%左右。其中还描述了γ-丁内酯法合成nmp反应在高温条件下分两步进行:第一步开环胺化生成γ-羟基丁酰胺,第二步缩合闭环反应生成nmp产品。反应式如下:
[0015][0016]
同时,还认为γ-丁内酯法中水的作用是双向的,一方面,水是副产物,会影响反应平衡,因此要控制水含量;另一方面,中间体链状γ-轻基丁酰胺脱水生成五元环应该是由非稳态转变为稳态的自发过程,但实际上脱水比胺化更困难,可能是能垒更大所致,而加入水可能降低了反应的活化能垒体现出对缩合闭环的促进作用,此外,大量水稀释了原料,在一定程度上起到了降低空速的作用,使得催化剂与原料接触时间更久,接触更为充分,促进了产物生成。
[0017]
引用文献7中介绍了采用微波辐射技术,将硅藻土作载体浸渍于磷钨酸制备成固载杂多酸催化剂,以γ-丁内酯和苯胺为原料合成了1-苯基-2-吡咯烷酮(npp),最佳条件为:磷钨酸负载量17.6%、催化剂用量1.5%(质量分数)、微波功率325w、辐射时间14min,γ-丁内酯与苯胺摩尔比1.0:1.25。在此条件下产率达96%以上,反应速率提高近10倍。
[0018]
综上所述,使用固体催化剂由内酯与有机胺化合物直接发生胺化反应进行内酰胺合成已经得到了应用。然而,也因为如此,目前的催化体系大多适用于有机胺的胺化反应,对于直接使用氨气由内酯合成酰胺的工艺仍然存在需要高温高压等苛刻条件的问题。
[0019]
引用文献:
[0020]
引用文献1:cn103547569a
[0021]
引用文献2:张玲钰等(参阅文献:张玲钰,王云川等.固体超强酸催化合成n-苯基吡咯烷酮的研究[j],2017第39卷第4期)
[0022]
引用文献3:cn105753768a
[0023]
引用文献4:cn1120151c
[0024]
引用文献5:cn1712396a
[0025]
引用文献6:沈宸等(参阅文献:沈宸,周维友等.zsm-5分子筛气相催化合成n-甲基吡咯烷酮[j],石油炼制与化工,2013年,44(1),51~55)
[0026]
引用文献7:胡珊玲等(参阅文献:张志德,林凤珍等赣南师范学院学报[j],2006第6期)
技术实现要素:
[0027]
发明要解决的问题
[0028]
如上所述,尽管已经尝试使用了各种固体催化剂、并使用有机胺化合物以使得内酯进行胺化反应而得到相应的内酰胺化合物。
[0029]
然而,从简化反应步骤的角度考虑,使用氨气而不是有机胺显然具有更好的工业生产前景。虽然例如引用文献3中,也提出了使用液氨的技术方案,但在使用液氨过程中,对于设备的要求很高,因此,反应条件并不能说是便利的。
[0030]
因此,鉴于以上问题,本发明所要解决的技术问题在于提供一种新的胺化反应方法,该方法能够避免上述内酯化合物的胺化反应存在的高温、高压操作且反应时间较长的
问题,同时也能够减轻对设备的选用、过程的操作和反应的控制要求。
[0031]
进一步,本发明也提供了一种用于上述胺化反应的催化剂,该催化剂特别能够高效催化(双环)内酯化合物与氨气进行胺化反应直接制备相应内酰胺。
[0032]
此外,本发明所要解决的技术问题也涉及对于过量反应物的处理,例如过量的氨如何回收套用的问题。
[0033]
用于解决问题的方案
[0034]
经过本发明发明人长期研究发现,通过如下技术方案的实施能够解决上述技术问题:
[0035]
[1].本发明首先提供了一种酯类化合物胺化的方法,其中,所述反应包括使用氨气在催化剂的存在下,将所述酯类化合物转变为酰胺化合物的步骤;
[0036]
其中,所述催化剂包括负载型催化剂,所述负载型催化剂包括载体以及活性组分,
[0037]
所述载体包括过渡金属的磷酸盐;
[0038]
所述活性组分包括金属含氧酸的铵盐。
[0039]
[2].根据[1]所述的方法,其中,所述酯类化合物包括内酯类化合物;所述负载型催化剂中,所述活性组分的含量以所述负载型催化剂的总质量计为15质量%~45质量%。
[0040]
[3].根据[1]或[2]所述的方法,其中,所述载体中磷酸盐中的过渡金属包括元素周期表第四周期中的过渡金属中的一种或多种。
[0041]
[4].根据[1]~[3]任一项所述的方法,其中,所述金属含氧酸包括v、nb、ta、mo或w元素的金属含氧酸盐中的一种或多种。
[0042]
[5].根据[1]~[4]任选一项所述的方法,其中,所述负载型催化剂的用量为酯类化合物的1质量%~50质量%。
[0043]
[6].根据权利要求1~5任选一项所述的方法,其中,所述氨气的用量为酯类化合物的1.1倍质量以上。
[0044]
[7].根据[1]~[6]任选一项所述的方法,其中,所述胺化反应是在水存在下进行的,所述水的用量为所述酯类化合物的10倍质量以下。
[0045]
[8].根据[1]~[7]任选一项所述的方法,其中,所述胺化反应的温度为50~180℃,压力为10atm以下。
[0046]
[9].根据[1]~[8]任选一项所述的方法,其中,所述胺化反应在固定床反应器中进行,催化剂的体积空速为0.1-2h-1
。
[0047]
[10].进一步,本发明也提供了一种用于酯类化合物胺化的催化剂,其中,所述催化剂为负载型催化剂,并且,在所述负载型催化剂的存在下,氨气与酯类化合物接触后可以将所述酯类化合物转变为酰胺化合物,
[0048]
所述负载型催化剂包括载体以及活性组分,
[0049]
所述载体包括过渡金属的磷酸盐;
[0050]
所述活性组分包括金属含氧酸的铵盐。
[0051]
[11].根据[10]所述的催化剂,其中,所述酯类化合物包括内酯类化合物;所述负载型催化剂中,所述活性组分的含量以所述负载型催化剂的总质量计为15质量%~45质量%。
[0052]
[12].根据[10]或[11]所述的催化剂,其中,所述磷酸盐包括磷酸铁;所述金属含
氧酸包括钨酸或钼酸中的一种或它们的混合物。
[0053]
发明的效果
[0054]
通过上述技术方案的实施,本发明能够获得如下的技术效果:
[0055]
1)本发明的制备负载型(杂多酸铵)催化剂比表面积大,催化剂活性优异,且活性组分与载体之间没有界面,高度一体化,活性组分不易流失,连续使用500h后,催化剂的性能仍保持不变。
[0056]
2)本发明的负载型催化剂,能够高效实现酯类化合物,尤其是内酯类化合物,例如双环内酯化合物与氨气的胺化反应。在这些反应中,本发明的胺化反应具有反应条件温和、反应时间短、催化剂易于回收利用、产品收率高等优点,适合大规模工业化生产。
[0057]
3)本发明以廉价的氨气作为胺源,相比较现有技术中采用有机胺或液氨为胺源制备6,6-二甲基-3-氮杂双环[3.1.0]己烷的方法,省去了脱苄步骤或者是降低了反应设备的要求,原子经济性显著提高。
[0058]
4)本发明中,未反应的氨可以方便的循环套用,使制备成本大大降低。
具体实施方式
[0059]
以下,针对本发明的内容进行详细说明。以下所记载的技术特征的说明基于本发明的代表性的实施方案、具体例子而进行,但本发明不限定于这些实施方案、具体例子。需要说明的是:
[0060]
本说明书中,使用“数值a~数值b”表示的数值范围是指包含端点数值a、b的范围。
[0061]
本说明书中,使用“以上”或“以下”表示的数值范围是指包含本数的数值范围。
[0062]
本说明书中,使用“可以”表示的含义包括了进行某种处理以及不进行某种处理两方面的含义。
[0063]
本说明书中,使用“任选”或“任选的”表示某些物质、组分、执行步骤、施加条件等因素使用或者不使用以及对使用方式没有限定。
[0064]
本说明书中,2019冠状病毒病(covid-19)及其病毒的命名采用世界卫生组织(who)的命名原则,即,造成2019冠状病毒病(covid-19)的病毒(此前称为“2019新型冠状病毒”)及其引发的疾病的正式名称已经公布。正式名称是:
[0065]
疾病:2019冠状病毒病(covid-19)
[0066]
病毒:严重急性呼吸综合征冠状病毒2(sars-cov-2)。
[0067]
本说明书中,所使用的单位名称均为国际标准单位名称,并且如果没有特别声明,所使用的“%”均表示重量或质量百分含量。
[0068]
本说明书中,所提及的“一些具体/优选的实施方案”、“另一些具体/优选的实施方案”、“实施方案”等是指所描述的与该实施方案有关的特定要素(例如,特征、结构、性质和/或特性)包括在此处所述的至少一种实施方案中,并且可存在于其它实施方案中或者可不存在于其它实施方案中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方案中。
[0069]
本发明首要的公开了一种针对酯类化物的胺化方法。本发明的方法中,使用氨气,并且在催化剂的存在下,将所述酯类化物胺化为酰胺化合物。以下将对上述胺化方法进行具体说明。
[0070]
(酯类化合物)
[0071]
对于本发明的胺化方法所针对的酯类化合物,原则上没有特别限制,可以是含羧基(或酰卤)化合物与含羟基化合物进行酯化反应而得到的任意的酯类化合物。
[0072]
在一些具体的实施方案中,这样的酯类化合物可以是一个分子中含有一个或一个以上酯键的化合物。另外,这样的酯类化合物可以通过酯键而成环。
[0073]
如上所述的,现有的研究通常认为在将酯基的氧进行胺化时,首先发生的是酰基和氧键的断裂,进而再通过脱水反应而形成酰胺结构,而从热力学角度,脱水形成最终的酰胺结构的势垒更大。
[0074]
因此可见,无论从反应热力学还是从分子动力学的角度考虑,本发明的胺化反应优选是针对通过酯键而成环的那些内酯类化物的胺化反应。
[0075]
对于所述内酯类化合物,其包括酯基的环上可以具有4个或4个以上的碳原子,优选地,所述内酯类化合物包括酯基的环上具有4~20个,进一步优选地,具有4~10个,更优选地,具有4~6个碳原子。
[0076]
此外,对于内酯类化物,从胺化效率的角度考虑,其环结构上包括1个或2个酯基,优选地,包括1个酯基。
[0077]
进一步,对于所述内酯类化合物,除了其包括含酯基的环以外,还可以通过对环上的碳原子的取代而引入任何需要的取代基团、功能性基团或者其他的药学上所要的结构。
[0078]
具体可以列举的可以用于本发明酯类化物包括以下结构a的内酯化合物:
[0079][0080]
其中,r表示在包括酯基的环上任意位置的取代基,其每次出现时,相同或不同,可以相互独立的表示氢原子、烃基、极性基团等。其中,所述极性基团优选为含卤素基团、含羧基的基团、含羟基基团、含酯基的基团或含氰基的基团等。另外,对于相邻的r之间,可以通过直接键或二价连接基团而形成环。
[0081]
另外,n表示0~6的整数,优选为0~3的整数。
[0082]
在本发明进一步优选的实施方案中,所述内酯类化合物包括如下通式(1)结构的化合物:
[0083][0084]
其中,r1表示氢原子、烃基或上文所定义的那些极性基团,优选的,可以为氢原子
或极性基团。
[0085]
(氨气)
[0086]
本发明通过使用氨气以完成对上述酯类化合物的胺化反应。其中,对于氨气的来源没有特别限制。例如,在胺化反应过程中,作为原料,可以直接使用氨气,也可以使用液态氨,并通过气化而使用。
[0087]
(催化剂)
[0088]
对于本发明的催化剂,主要用于促进胺化反应的完成。
[0089]
具体而言,本发明所述催化剂包括负载型催化剂,所述负载型催化剂包括载体以及活性组分。
[0090]
载体
[0091]
作为本发明负载型催化剂的载体,可以通过过渡金属的磷酸盐而形成。
[0092]
在本发明一些具体的实施方案中,所述过渡金属可以选自元素周期表的第四周期中的过渡金属元素中的一种或多种,更优选地,选自第四周期中viii族中的金属元素中的一种或多种,最优选地,所述过渡金属元素选自铁。
[0093]
本发明中,对于载体的形成方法,原则上没有特别限制,在一些优选的实施方案中,可以通过共沉淀-焙烧的方法而得到所述载体。
[0094]
在所述共沉淀的步骤中,主要是通过将磷源和铁源形成于混合溶液中以形成沉淀物而得到。
[0095]
其中,对于本发明可用磷源没有特别限制,可以包括磷酸、磷酸一氢金属盐、磷酸二氢金属盐、磷酸一氢氨、磷酸二氢铵、磷酸金属盐、磷酸铵中的一种或几种;另外,对于铁源,也没有特别的限制,本发明可用的铁源包括金属铁粉、硝酸铁、硝酸亚铁、硫酸亚铁、硫酸铁、氧化亚铁、氧化铁、氯化亚铁或氯化铁中的一种或几种。
[0096]
通常可以预先将磷源或铁源中的至少一种形成溶液,进而进行混合。对于进行共沉淀时,铁源与磷源的摩尔比(以铁元素与磷元素摩尔比计)通常可以为0.1~0.5:1,优选地,铁源与磷源的摩尔比可以为0.2~0.4:1。
[0097]
此外,在进行混合磷源与铁源的混合时,可以根据需要而使用各种助剂。在本发明一些具体的实施方案中,所述助剂可以包括氧化剂、ph调节剂、模板成分等。
[0098]
在得到沉淀后,从进一步得到有利的沉淀形貌而言,在本发明一些具体的实施方案中可以将共沉淀步骤得到的沉淀物置于含有表面活性剂的溶液体系中分散,并与醇类溶剂进行混合后,进行熟化处理。对于所述表面活性剂,在本发明一些具体的实施方案中,可以选自离子型表面活性剂、非离子表面活性剂中的一种或多种,典型可以列举的表面活性剂包括:烷基溴化铵、泊洛沙姆、聚乙二醇、聚乙烯吡咯烷酮、十二烷基苯磺酸钠、脂肪醇聚氧乙烯醚等。此外,任选的,上述分散中,必要时也可以使用络合剂(例如乙二胺四乙酸)等助剂。
[0099]
所述熟化处理的温度可以为200℃以下,优选为100℃~160℃,熟化时间没有特别限制,通常可以控制在5~30h,优选为8~24h之间。
[0100]
通过熟化的沉淀物进一步通过清洗、干燥和焙烧以得到最终的载体。在本发明一些具体的实施方案中,可以将清洗、干燥后的沉淀物在400~600℃条件下焙烧2~6h。
[0101]
对于最终得到的催化剂载体的固体形貌,可以为颗粒状、粉末状或它们的混合物,
对于颗粒或者粉末,优选地,可以具有球形或接近球形的外表,并且具有表面多孔结构。
[0102]
对于本发明的典型的载体的形成过程,可以为如下:
[0103]
先将铁源和磷酸根源分别溶于去离子水中得到溶液a和溶液b,混合溶液a和溶液b得到沉淀物,过滤取出沉淀物,然后将沉淀物悬浮于表面活性剂中,将得到的悬浮液与乙醇混合置于高压反应器中在100~200℃下反应8~30h,过滤、洗涤、烘干后得到固体产物,之后将得到的固体产物在400~600℃下焙烧2~6小时,得到多孔的磷酸铁化合物。
[0104]
活性组分
[0105]
本发明中,所述催化剂中的活性组分包括金属含氧酸的铵盐。
[0106]
对于金属的含氧酸,可以选自含有v、nb、ta、mo或w元素的金属含氧酸盐中的一种或多种。并且,在本发明一些优选的实施方案中,从脱水效果考虑,所述金属的含氧酸可以选自钨酸、钼酸中的一种或者它们的混合。
[0107]
对于所述金属含氧酸的铵盐的合成方法,没有特别限定,可以使用本领域常规的制备工艺而得到。
[0108]
负载型催化剂的形成
[0109]
本发明中,通过将上述的活性组分负载于所述载体以得到本发明的负载型催化剂。更具体而言,通过活性组分的负载以及焙烧等工序而得到最终的催化剂。
[0110]
对于本发明的活性组分的负载方法,原则上没有特别限制,可以使用的方法,包括干混或湿混等混合方式。
[0111]
在本发明一些优选的实施方案中,可以使用将载体固体物放入含有活性组分的分散液或溶液中,优选地,在酸性条件下进行混合以得到负载型催化剂前体。
[0112]
进一步,将所述前体进行焙烧处理。在本发明一些具体的实施方案中,所述焙烧处理的温度可以为200~400℃,优选为300~360℃,焙烧时间可以为2~6h。
[0113]
对于本发明的典型的负载型催化剂的形成过程,可以为如下:
[0114]
将金属铵盐溶于去离子水中,加入制备好的多孔磷酸铁固体,调ph至1~3,室温下搅拌8~16h,过滤得到前驱体。之后将得到的前驱体在200~400℃下加热2~6h,得到负载型催化剂。
[0115]
对于本发明的催化剂的组成,在一些优选的实施方案中,以所述负载型催化剂的总质量计,活性组分的含量为15~45质量%,优选为20~40质量%。
[0116]
进一步,对于本发明的负载型催化剂,具有高的比表面积,在一些具体的实施方案中,本发明的负载型催化剂具有根据bet测试方法得到的100-160m2/g的比表面积。
[0117]
其他可用的催化剂成分
[0118]
本发明中,在胺化反应中,除了使用以上所描述的催化剂以外,在需要是,尤其是针对胺化反应中的脱水形成酰胺的步骤,还可以使用其他利于脱水的催化剂。
[0119]
对于这些其他的催化剂,主要可以包括其他的杂多酸铵型催化剂,在一些具体的实施方案中,这些其他的杂多酸铵型催化剂可以包括活性组分、助剂及载体,活性组分为杂多酸铵盐见式(s),助剂为氧化镍,载体为氧化硅;以催化剂的重量为基准,杂多酸铵盐的含量为5%~45%,优选为10%~40%,助剂以氧化物计的含量为3%~18%,优选为5%~15%,载体的含量为37%~92%,优选为45%~85%;
[0120]hm
(nh4)nyx
12o40
(s)
[0121]
其中x代表w或mo,y代表si或p;当y代表si时,m n=4,n取值为0.1~1.0;当y代表p时,m n=3,n取值为0.1~1.0。
[0122]
所述氧化硅载体的性质如下:比表面积为500~820m2/g,孔容为0.62~0.92ml/g,平均孔直径为4.6~6.6nm。
[0123]
对于这些其他的催化剂成分的用量没有特别限制,在一些具体的实施方案中,以本发明胺化反应中使用的催化剂的总量计,上述其他的催化剂成分的用量可以为30质量%以下,优选为20质量%以下,进一步优选为10质量%以下。
[0124]
(胺化反应)
[0125]
本发明的胺化反应是在上述催化剂的存在下,将酯类化合物与氨气接触,进而得到酰胺化合物的反应过程。
[0126]
在一些优选的实施方案中,从便于内酯的开环反应的角度考虑,所述胺化反应中,还使用水。
[0127]
在一些具体的胺化反应过程中,在水和催化剂的存在下,酯类化合物的酰-氧键打开,一部分形成-oh基团,一部分形成h2n-co-基团。进一步,在上述催化剂的作用下,上述两部分进一步脱水以形成酰胺键。
[0128]
对于胺化反应中上述负载型催化剂的用量,在本发明一些具体的实施方案中,本发明的负载型催化剂的用量为可以为酯类化合物的1质量%~50质量%,优选为2质量%~40质量%。
[0129]
对于胺化反应中,如果使用了水,则水的用量可以为酯类化物的摩尔量的5~10倍,优选为6~9倍,如果水的用量过大,则对于胺化反应形成酰胺过程中的脱水而言是不利的。
[0130]
进一步,对于胺化反应中的反应温度,通常可以为50~180℃,优选地,可以为80~170℃,进一步优选地可以为100~160℃。对于胺化反应的压力,由于使用的是氨气,因此,从有利于胺化反应进行以及减少设备负担的角度考虑,胺化反应的压力可以控制在常压或以上,且10atm以下,优选的控制在6atm以下。对于胺化反应中氨气的用量,可以适当的过量使用氨气以促进胺化反应效率,在一些具体的实施方案中,所述氨气的用量为酯类化合物的1.1倍质量以上,例如1.1~2倍,优选为1.2~1.6倍。
[0131]
对于胺化反应中,酯类化合物的停留时间,通常可以为0.1~8h,优选为0.5~6h。
[0132]
另外,对于胺化反应中使用的氨气,可以在反应结束后,对体系中未反应的氨气进行回收,并供给给下一次的胺化反应。
[0133]
此外,对于所述胺化反应的装置,原则上没有特别限制。在本发明一些优选的实施方案中,可以使用固定床反应装置,固定床反应装置中,催化剂的体积空速可以控制为0.1-2h-1
。
[0134]
在本发明一些具体的实施方案中,所述固定床反应器中催化剂装填在反应器中段,反应器两端装填石英砂支撑,原料液从反应器底部进入,氨气从反应器顶部进入,在上部石英砂段被预热,反应在催化剂床层进行,反应器外部有加热和保温装置,通过热电偶测温、控温。催化剂装填量为固定床反应器总体积的20-30%。
[0135]
通过上述胺化反应,可以将酯类化合物尤其是可以将通式(1)的化合物转化成通式(2)的化合物,优选地,可以通过便利且相对温和的条件而得到式(2a)的化合物:
[0136][0137]
进一步,通过对式(2a)的还原可以得到式(3)所表示的化合物,而式(3)的化合物可以进一步作为医药合成中间体。
[0138]
因此,基于本发明的上述所提供的新的胺化方法,可以高效率且条件温和的方式提供相关药物中间体的前体的生产,因此,本发明的胺化方法也可以作为药物合成中的中间合成步骤。
[0139]
此外,通过本发明胺化方法以及后续还原方法所能够提供的药物中间体,可以进一步用于包括丙肝蛋白酶抑制剂类药物化合物或治疗新型冠状病毒(sars-cov-2)的药物化合物的制备。
[0140]
实施例
[0141]
以下将通过具体的实施例对本发明做出进一步的说明:
[0142]
制备例(催化剂的制备)
[0143]
制备例1
[0144]
(1)载体磷酸铁的制备
[0145]
将4g硝酸铁溶于20ml去离子水中,另将9g磷酸一氢钠溶于60ml去离子水中。混合上述两溶液得到沉淀物,过滤取出沉淀物,然后将沉淀物悬浮于10ml的十二烷基硫酸钠中,将得到的悬浮液与乙醇混合置于聚四氟乙烯内衬高压反应器中在150℃下反应24小时,过滤、洗涤、烘干后得到固体产物,之后将得到的固体产物在500℃焙烧4小时,得到多孔的磷酸铁化合物。
[0146]
(2)杂多酸催化剂的制备
[0147]
将37.5g钼酸铵溶于100ml去离子水,加入制备好的100g多孔磷酸铁固体,用hcl调ph至2,室温下搅拌12h,过滤得到前驱体。之后将得到的前驱体在300℃下加热4h,得到杂多酸催化剂,比表面积为138m2/g,钼酸铵负载量16.8%。
[0148]
制备例2
[0149]
(1)载体磷酸铁的制备
[0150]
将4g硝酸铁溶于20ml去离子水中,另将9g磷酸一氢钠溶于60ml去离子水中。混合上述两溶液得到沉淀物,过滤取出沉淀物,然后将沉淀物悬浮于10ml的十二烷基硫酸钠中,将得到的悬浮液与乙醇混合置于聚四氟乙烯内衬高压反应器中在160℃下反应18小时,过滤、洗涤、烘干后得到固体产物,之后将得到的固体产物在520℃焙烧3小时,得到多孔的磷酸铁化合物。
[0151]
(2)杂多酸催化剂的制备
[0152]
将120g钼酸铵溶于315ml去离子水,加入制备好的100g多孔磷酸铁固体,用hcl调
ph至2,室温下搅拌12h,过滤得到前驱体。之后将得到的前驱体在300℃下加热4h,得到杂多酸催化剂,比表面积为143m2/g,钼酸铵负载量42.3%。
[0153]
制备例3
[0154]
(1)载体磷酸铁的制备
[0155]
将4g硝酸铁溶于20ml去离子水中,另将9g磷酸一氢钠溶于60ml去离子水中。混合上述两溶液得到沉淀物,过滤取出沉淀物,然后将沉淀物悬浮于10ml的十二烷基硫酸钠中,将得到的悬浮液与乙醇混合置于聚四氟乙烯内衬高压反应器中在160℃下反应20小时,过滤、洗涤、烘干后得到固体产物,之后将得到的固体产物在520℃焙烧3小时,得到多孔的磷酸铁化合物。
[0156]
(2)杂多酸催化剂的制备
[0157]
将90g钼酸铵溶于250ml去离子水,加入制备好的100g多孔磷酸铁固体,用hcl调ph至2,室温下搅拌12h,过滤得到前驱体。之后将得到的前驱体在300℃下加热4h,得到杂多酸催化剂,比表面积为147m2/g,钼酸铵负载量32.7%。
[0158]
制备例4
[0159]
(1)载体磷酸铁的制备
[0160]
将4g硝酸铁溶于20ml去离子水中,另将9g磷酸一氢钠溶于60ml去离子水中。混合上述两溶液得到沉淀物,过滤取出沉淀物,然后将沉淀物悬浮于10ml的十二烷基硫酸钠中,将得到的悬浮液与乙醇混合置于聚四氟乙烯内衬高压反应器中在160℃下反应20小时,过滤、洗涤、烘干后得到固体产物,之后将得到的固体产物在520℃焙烧3小时,得到多孔的磷酸铁化合物。
[0161]
(2)杂多酸催化剂的制备
[0162]
将90g钨酸铵溶于35ml去离子水,加入制备好的100g多孔磷酸铁固体,用hcl调ph至2,室温下搅拌12h,过滤得到前驱体。之后将得到的前驱体在300℃下加热4h,得到杂多酸催化剂,比表面积为144m2/g,钨酸铵负载量30.3%。
[0163]
制备例5
[0164]
(1)载体磷酸铁的制备
[0165]
将4g硝酸铁溶于20ml去离子水中,另将9g磷酸一氢钠溶于60ml去离子水中。混合上述两溶液得到沉淀物,过滤取出沉淀物,然后将沉淀物悬浮于10ml的十二烷基硫酸钠中,将得到的悬浮液与乙醇混合置于聚四氟乙烯内衬高压反应器中在160℃下反应20小时,过滤、洗涤、烘干后得到固体产物,之后将得到的固体产物在520℃焙烧3小时,得到多孔的磷酸铁化合物。
[0166]
(2)杂多酸催化剂的制备
[0167]
将30g钼酸铵溶于90ml去离子水,加入制备好的100g多孔磷酸铁固体,用hcl调ph至2,室温下搅拌12h,过滤得到前驱体。之后将得到的前驱体在300℃下加热4h,得到杂多酸催化剂,比表面积为146m2/g,钼酸铵负载量14.2%。
[0168]
实施例(用制备例制备到的催化剂用于胺化反应)
[0169]
实施例1
[0170]
(用制备例1制备到的催化剂用于胺化反应)
[0171]
将100ml上述杂多酸催化剂装入固定床中,催化剂的空速为0.5h-1
,内酯进料量为
1.48ml/min,氨的摩尔量为内酯摩尔量的2倍,助剂水的摩尔量为内酯摩尔量的5.7倍,内酯、水及氨气流经催化剂床层,催化剂床层温度为150℃,压力为6mpa,反应后取样,此条件下的内酰胺收率能达到90.5%。且经过500h的连续反应后,催化剂的性能保持不变,内酰胺收率能维持在90.5%。
[0172]
实施例2
[0173]
(用制备例2制备到的催化剂用于胺化反应)
[0174]
将100ml上述杂多酸催化剂装入固定床中,催化剂的空速为0.5h-1
,内酯进料量为1.48ml/min,氨的摩尔量为内酯摩尔量的2倍,助剂水的摩尔量为内酯摩尔量的5.7倍,内酯、水及氨气流经催化剂床层,催化剂床层温度为150℃,压力为6mpa,反应后取样,此条件下的内酰胺收率能达到97.3%。且经过500h的连续反应后,催化剂的性能保持不变,内酰胺收率能维持在97.3%。
[0175]
实施例3
[0176]
(用制备例3制备到的催化剂用于胺化反应)
[0177]
将100ml上述杂多酸催化剂装入固定床中,催化剂的空速为0.5h-1
,内酯进料量为1.48ml/min,氨的摩尔量为内酯摩尔量的2倍,助剂水的摩尔量为内酯摩尔量的5.7倍,内酯、水及氨气流经催化剂床层,催化剂床层温度为150℃,压力为6mpa,反应后取样,此条件下的内酰胺收率能达到94.8%。且经过500h的连续反应后,催化剂的性能保持不变,内酰胺收率能维持在94.8%。
[0178]
实施例4
[0179]
(用制备例2制备到的催化剂用于胺化反应)
[0180]
将100ml上述杂多酸催化剂装入固定床中,催化剂的空速为0.2h-1
,内酯进料量为1.48ml/min,氨的摩尔量为内酯摩尔量的2倍,助剂水的摩尔量为内酯摩尔量的5.7倍,内酯、水及氨气流经催化剂床层,催化剂床层温度为150℃,压力为6mpa,反应后取样,此条件下的内酰胺收率能达到98.1%。且经过500h的连续反应后,催化剂的性能保持不变,内酰胺收率能维持在98.1%。
[0181]
实施例5
[0182]
(用制备例4制备到的催化剂用于胺化反应)
[0183]
将100ml上述杂多酸催化剂装入固定床中,催化剂的空速为0.2h-1
,内酯进料量为1.48ml/min,氨的摩尔量为内酯摩尔量的2倍,助剂水的摩尔量为内酯摩尔量的5.7倍,内酯、水及氨气流经催化剂床层,催化剂床层温度为150℃,压力为6mpa,反应后取样,此条件下的内酰胺收率能达到95.1%。且经过500h的连续反应后,催化剂的性能保持不变,内酰胺收率能维持在95.1%。
[0184]
参考实施例
[0185]
(用制备例5制备到的催化剂用于胺化反应)
[0186]
将100ml上述杂多酸催化剂装入固定床中,催化剂的空速为0.5h-1
,内酯进料量为1.48ml/min,氨的摩尔量为内酯摩尔量的2倍,助剂水的摩尔量为内酯摩尔量的5.7倍,内酯、水及氨气流经催化剂床层,催化剂床层温度为150℃,压力为6mpa,反应后取样,此条件下的内酰胺收率能达到83.3%。且经过500h的连续反应后,催化剂的性能保持不变,内酰胺收率能维持在83.3%,显示,当催化剂的负载量低于本发明优选的范围时,有可能使得反应
收率有降低的趋势。
[0187]
需要说明的是,尽管以具体实例介绍了本发明的技术方案,但本领域技术人员能够理解,本公开应不限于此。
[0188]
以上已经描述了本公开的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。本文中所用术语的选择,旨在最好地解释各实施例的原理、实际应用或对市场中的技术的改进,或者使本技术领域的其它普通技术人员能理解本文披露的各实施例。
[0189]
产业上的可利用性
[0190]
本发明提供的合成方法可以在工业上用于制备医药中间体化合物。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。