一种能够实现单个aav病毒包被的迷你碱基编辑器
技术领域
1.本发明公开了一种能够实现单个aav病毒包被的迷你碱基编辑器,包括tada-8e片段和spcas9缺失变体片段,开发出编辑窗口大的miniabe以及编辑窗口小的miniabe*,能够实现单个aav病毒包装,更利于基因治疗,具有良好的应用前景,属于生物技术领域。
背景技术:
2.crispr/cas9技术在目标位点引入dna双链断裂(dsb)对生物医药领域起到革命性作用,通过修改或纠正有害基因来促进遗传病的治疗。在sgrna引导下,cas9蛋白能够到达指定区域并发挥核酸酶活性进行dna双链切割。但直接使用cas9系统往往难以实现精确编辑。基于crispr技术上开发出更为精确的单碱基编辑系统(abe和cbe),可以实现四种碱基类型的转换(c-t,g-a,a-g和t-c)。已知的疾病相关的基因突变中有一半是点突变,原则上近一半的致病位点突变可以通过将a
·
t碱基对转化成g
·
c碱基对来纠正,因此abes对于致病等位基因的纠正特别有用。但最常用的spcas9有1368个氨基酸组成,4104 bp。基于spcas9的abes及其sgrna远远超过了aav载体的限制。腺相关病毒(aav)是体内给药的最佳载体,但aav的有效载荷能力有限(一般小于5kb)。
3.为了绕过aav的有效荷载限制,将spcas9拆成两部分并与sgrna表达单元一起包装成两个独立的aav,通过两个aav中的反向末端重复(itr)序列的重组反应或内含子介导的蛋白质反式剪接从而允许全长的spcas9在体内重新组装。然而,与单一载体系统相比,双载体系统通常需要更高的病毒剂量,生产成本更高,难以按化学计量学进行滴定,而且操作起来也很麻烦。另一种途径是开发尺寸更小的cas9,如sacas9(3.2 kb)、casx(3kb)以及cjcas9等。但结构紧凑的cas9效率不如spcas9,且具有更加严格的pam。因此开发基于spcas9的abe载体大小缩小到aav载体限制,有助于我们对碱基编辑器的应用,特别是在医疗领域对相关疾病进行精准基因治疗方面。
技术实现要素:
4.本发明目的在于提供一种碱基编辑工具及其用途,尤其是公开一种能够实现单个aav病毒包被的迷你碱基编辑器,解决了spcas9的abe系统无法实现单一aav包被的技术缺点。
5.本发明公开了一种能够实现单个aav病毒包被的迷你碱基编辑器,包括tada-8e和spcas9缺失变体的两种迷你碱基编辑器;其中,tada-8e与spcas9缺失rec2结构域第144-296位氨基酸以及hnh结构第786-855位氨基酸构建的载体命名为miniabe,核苷酸序列如seqidno.1tada-8e与spcas9缺失rec2结构域第144-296位氨基酸,hnh结构第762-855位氨基酸以及tada-8e与spcas9的连接linker构建的载体命名为miniabe*,核苷酸序列如seq id no.2。
6.本发明所述的一种能够实现单个aav病毒包被的迷你碱基编辑器,其特征在于:
miniabe-p5小鼠肝脏编辑效率图)。
具体实施方式
13.以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域人员所熟知的常规手段。下述实施例中所使用的材料、试剂等如无特殊说明,均可从商业途径得到。
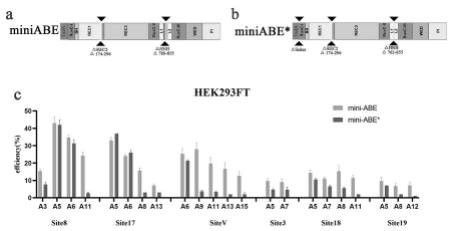
14.实施例1miniabe载体构建及效率验证1.1miniabe载体构建pcr将addgene来源的abe8e(#138489)使用nebq5高保真聚合酶pcr成两个片段,引物为:fragment1f:aacatcgtgatcgaacggcagatcacaaagcacg;fragment1r:aaattttgctcgcgcactactcagcga;vector1f:gcgcgagcaaaatttaagctacaaca;vector1r:tcgatcacgatgttctcgggc;之后将pcr产物进行凝胶电泳,切割fragment14050,vector13708条带,使用天根凝胶回收试剂盒进行胶回收;将胶回收片段1:1使用诺唯赞无缝克隆试剂盒进行重组,重组程序为50℃30min。随后将重组产物使用天根dh5α感受态细胞进行转化,过程为(冰浴30min,热激90s,冰浴3min,涂板)。得到单克隆以后挑菌测序,得到克隆1,随后将克隆1继续用fragment2f:atcaagttccggggcctgagcgcctctatgatcaag;fragment2r:gcgccacgcttcccgaaggagaaagg;vector2f:cgggaagcgtggcgc;vector2r:gccccggaacttgatcatgtg;进行pcr,胶回收,重组得到miniabe(图1a),测序正确后采用天根大提试剂盒进行质粒提取,miniabe序列如seqidno.1。
15.1.2sgrna载体构建选择6个内源基因位点,根据目标序列设计sgrna并合成oligos,所用到的sgrna序列如表1所示。在每个sgrna的上游序列5’端加cacc序列接头,下游序列的5’加aaac序列接头。经合成以后,上下游通过程序(95℃5min室温冷却30min)退火,将退火产物连接到进bbsi酶切的74707sgrna骨架载体,随后经过转化,挑单克隆以及测序得到正确的sgrna,并通过大提得到高纯度质粒。
16.表1.hek293ft细胞sgrna序列sgrnaoligo-foligo-rsite8caccgaaataatgccatcttccgctaaacagcggaagatggcattatttcsite17caccgtgtaagacctcaaaagcacaaacgtgcttttgaggtcttacacsitevcaccgtgtgcaccagacataaataaaaacttatttatgtctggtgcacacsite3caccgaacacaaagcatagactgcaaacgcagtctatgctttgtgttcsite18caccgatgagaaggagaagttcttaaacaagaacttctccttctcatc
site19caccgaatactaagcatagactccaaacggagtctatgcttagtattc1.3细胞培养与转染将hek293ft细胞接种于10% fbs的dmem培养基中,其中含有1%双抗,在含有5% co2的37℃细胞培养箱中进行培养。用于转染的细胞,前一天接种于6孔板中进行培养,第二天观察细胞,当细胞密度为75%左右时,用含10% fbs,不含双抗的培养基换液,使用hiefftranstm liposomal transfection reagent进行转染,转染时6孔板每孔的量分别是miniabe 2μg,sgrna 1μg。将质粒混合后用250ulopti-mem稀释并混匀作为试剂a。同时,将9ul的hieff转染试剂与250μl的opti-mem稀释并混匀作为试剂b,静止5min。将上述试剂a与b混合,吹打混匀,静止20min后逐滴加入待转染的6孔板细胞中,放回37℃培养箱培养,12h后换一次新鲜培养基;48h后收集细胞,然后用细胞裂解液裂解(诺唯赞one step mouse genotyping kit)并鉴定基因型。
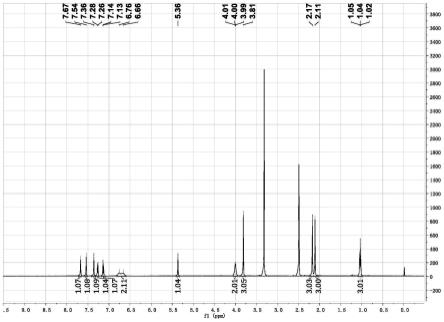
17.1.4 miniabe在内源基因位点编辑效率检测根据实验需求设计引物,将上述裂解产物为模板,对靶位点附近序列进行pcr扩增,将扩增产物凝胶电泳检测目的条带后,送上海生工公司进行一代测序。用于目标位点pcr扩增体系如下:2
×
taq(天根)12.5μl,f(10pmol/μl)1μl,r(10pmol/μl)1μl,模板1μl,ddh2o补齐到25μl。测序结束后将测序结果用editr分析网站进行效率分析,检测结果如图1c所示。
18.实施例2miniabe载体构建及效率验证2.1 miniabe*载体构建与方法1.1所述,将miniabe使用;fragment3f:cagagctccatcaacgacaagaagtacagcatcggcc;franment3r:cttatcagctccctcgtcacctcccag;vector3f:gagggagctgataagcgcac;vector3r:gttgatggagctctgggcct;进行pcr扩增,胶回收重组,转化,测序得到克隆2,再将克隆2使用:fragment4f:aacagccgcgagagaggcggcctgagc;fragment4r:gagctgaatgaagccataccaaacgac;vector4f:ggcttcattcagctccggttc;vector4r:tctctcgcggctgttcttctg;进行pcr扩增,胶回收,重组,转化,测序,大提质粒得到miniabe*(图1b),序列如seqidno.2所示。
19.2.2 细胞培养与转染与1.3所述一致。
20.2.3 miniabe*在内源性基因位点编辑效率检测与1.4所述一致,结果如图1c所示。
21.2.4 miniabe与miniabe*编辑窗口对比按照1.2所述,构建含有不同a的sgrna,序列如表2所示,按1.3,1.4转染细胞,并评估编辑效率,统计a3-a15的编辑效率,得到miniabe和miniabe*的大致窗口范围,如图2所
示。
22.表2.窗口评估细胞sgrna序列sgrnaoligo-foligo-rsite-atmcaccgtacctgaatgattcctgccaaacggcaggaatcattcaggtacsite-mtm1caccgttattctccaatggtgattaaacaatcaccattggagaataacsite-serpina1caccggccattgccggtggtcagcaaacgctgaccaccggcaatggccsite-thcaccgcgcctcacccttgggccccgaaaccggggcccaagggtgaggcgcsite-scnn1gcaccgtgctgtacctgccaaggtgaaaccaccttggcaggtacagcacsite-lipacacccttcctgcaacatggcttgcaaacgcaagccatgttgcaggaag实施例3aav载体的构建以及效率评估3.1 aav-miniabe载体构建由金斯瑞生物公司合成minicmv以及短poly(a)替换miniabe中的cmv启动子及bgh pa序列得到克隆3,并将u6启动子驱动的sgrna序列重组在克隆3上得到克隆4.之后使用:fragment5f:ggtacaattcacgcggactcacggggatttccaag;fragment5r:ggggttcctgcggccaaaaaaagcaccgactcggt;pcr扩增克隆4并连接到经mlui和noti酶切的aav骨架载体上,转化,测序,提取dna后得到aav-miniabe如图3a上所示,序列见seqidno.3。
23.3.2 aav-miniabe*载体构建由金斯瑞生物公司合成minicmv以及短poly(a)序列替换miniabe*中的cmv启动子及bgh pa序列得到克隆5,并将u6启动子驱动的sgrna序列重组在克隆3上得到克隆6.之后使用:fragment5f:ggtacaattcacgcggactcacggggatttccaag;fragment5r:ggggttcctgcggccaaaaaaagcaccgactcggt;pcr扩增克隆6并连接到经mlui和noti酶切的aav骨架载体上,转化,测序,提取dna后得到aav-miniabe*如图3a下所示,序列见seqidno.4。
24.3.3 pcsk9sgrna构建选择5个小鼠pcsk9基因位点,根据目标序列设计sgrna并合成oligos,所用到的sgrna序列如表3所示。按照1.2所述将sgrna连接到bbsi酶切后的aav-miniabe和aav-miniabe*载体中,随后通过dh5α转化,挑菌,测序得到正确连接的sgrna,并通过大提质粒得到高纯度的质粒。
25.表3. n2a细胞sgrna序列sgrnaoligo-foligo-rp4caccgttctaggctgcagcttccataaacatggaagctgcagcctagaacp5caccgttgcaggcctggagtttattaaacaataaactccaggcctgcaacp6caccgactcctacctgtgagaacaaaactgttctcacaggtaggagtcp7caccgtctaggctgcagcttccattaaacaatggaagctgcagcctagacp10caccggaagatggaagcagccaggaaaccctggctgcttccatcttcc3.4 细胞培养与转染
将n2a细胞接种于10�s的dmem培养基中,其中含有1%双抗,在含有5%co2的37℃细胞培养箱中进行培养。用于转染的细胞,前一天接种于6孔板中进行培养,第二天观察细胞,当细胞密度为75%左右时,用含10�s,不含双抗的培养基换液,使用hiefftranstmliposomaltransfectionreagent进行转染,转染时6孔板每孔的量分别是aav载体3μg。将质粒混合后用250ulopti-mem稀释并混匀作为试剂a。同时,将9ul的hieff转染试剂与250μl的opti-mem稀释并混匀作为试剂b,静止5min。将上述试剂a与b混合,吹打混匀,静止20min后逐滴加入待转染的6孔板细胞中,放回37℃培养箱培养,12h后换一次新鲜培养基。48h后收集细胞,然后用细胞裂解液裂解(诺唯赞onestepmousegenotypingkit)并鉴定基因型。
26.3.4aav载体在内源基因位点编辑效率检测方法通1.4所述,经过比较发现p5位点效率最高如图3b所示,因此将上述aav-miniabe-p5与aav-miniabe*-p5的两种载体进行aav包装。
27.实施例4aav-miniabe-p5和aav-miniabe*-p5aav包装将aav-miniabe-p5送至派真生物公司进行包装,使用qpcr检测滴度,使用sds-page检测纯度图4a。将aav-miniabe*-p5送至吉满生物公司进行aav包被,使用sds-page检测纯度图4b,同样检测滴度。
28.包装滴度分别如表4:表4.aav生产滴度aav-miniabe-p52.86e 13gc/mlaav-miniabe-p53.19e 13gc/mlaav-miniabe*-p52.17e 13gc/ml实施例5aav-miniabe-p5小鼠体内验证5.1aav病毒注射将aav包被的miniabe病毒颗粒置于冰上解冻,用胰岛素注射针吸取病毒,尾静脉注射8周龄的雌鼠三只,每只的注射量大约为4e 12gcs的病毒,同样给与对照组小鼠注射相同的量的pbs,如图5a。
29.5.2编辑效率检测1个月后,将小鼠处死,取肝脏组织进行基因组提取,随后将目的基因pcr扩增,pcr引物加深度测序接头,将产物送杭州水稻所进行深度测序,计算突变比例如图5b所示。
30.综上所述,本发明有效克服了现有技术无法满足aav包装的缺点,制备了两个窗口互补的迷你碱基编辑器,并成功用于aav的包装,并将aav注射到小鼠体内后产生一定编辑效果,上述迷你碱基编辑器具有高度产业利用价值。
31.上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。