1.本发明属于有机合成和原料药的制备技术领域,具体地说是涉及一种非奈利酮原料药的制备方法。
背景技术:
2.非奈利酮(finerenone,bay 94-8862)是一种非甾体选择性盐皮质激素受体拮抗剂,在临床前研究中显示可阻断盐皮质激素受体过度激活导致的有害影响。在糖尿病患者中,盐皮质激素受体过度激活被认为会导致慢性肾病进展和心血管受损,这可能由代谢、血流动力学或炎症和纤维化等因素驱动。
3.2021年7月,基于fidelio-dkd iii期临床研究在慢性肾病伴2型糖尿病成人患者中的阳性结果,美国fda批准非奈利酮(finerenone,)上市,目前非奈利酮已经在中国和全球多个其他国家地区递交上市申请,正在进行审评。
4.2021年12月,欧洲药品管理局(ema)人用医药产品委员会(chmp)推荐批准非甾体选择性盐皮质激素受体拮抗剂非奈利酮的上市申请,推荐非奈利酮(10mg或20mg)用于慢性肾病(3和4期并伴有白蛋白尿)伴2型糖尿病成人患者的治疗,非奈利酮一旦获得批准,将成为第一种用于改善慢性肾病伴2型糖尿病成年患者肾脏预后的非甾体选择性盐皮质激素受体拮抗剂。
5.非奈利酮化学结构如下所示:
[0006][0007]
目前,专利us2018244668a1公开了非奈利酮(finerenone)的制备方法,使用3-氧代丁酸2-氰乙基酯为原料,其合成路线如下:
[0008][0009]
目前工艺主要存在如下几方面问题:1)由于使用的3-氧代丁酸2-氰乙基酯不常见,原料的来源少,使得制备有一定的局限性;2)第二步采用高压反应,危险系数较高,不利于放大生产;3)最后的拆分步骤损失大等问题。
技术实现要素:
[0010]
为了克服现有技术存在的不足,本发明提供了一种非奈利酮原料药的制备方法。
[0011]
本发明所采用的技术方案为:
[0012]
一种非奈利酮原料药的制备方法,包括下述步骤:
[0013]
(1)4-甲酰基-3-甲氧基苯甲腈和乙酰乙酸脂类化合物,经过偶联得到结构式如(i)所示的化合物1;
[0014][0015]
(2)化合物1和4-氨基-5-甲基吡啶-2-醇经过脱水环合,得到结构式如(ii)所示的化合物2;
[0016][0017]
(3)化合物2经过原甲酸乙酯乙基化,得到结构式如(iii)所示的化合物3;
[0018][0019]
(4)化合物3在碱性条件下水解,得到结构式如(iv)所示的化合物4;
[0020][0021]
(5)化合物4经过cdi活化,氨解得到结构式如(v)所示的化合物5;
[0022][0023]
(6)化合物5在催化剂作用下回流反应,最后经过酒石酸拆分,得到非奈利酮原料药。
[0024]
其合成路线为:
[0025][0026]
作为优选,步骤(6)中所述催化剂为s2o
82-/zro2/γ-al2o3。本发明采用的催化剂经实验验证可以提高原料药构型比例。
[0027]
作为优选,所述催化剂通过下述方法制备得到:称取50g的八水合氧氯化锆,加450g的去离子水配制成质量分数为10%的溶液,在匀速搅拌的状态下,慢慢滴加氨水,至ph到10,停止加氨水此时生成白色沉淀zr(oh)4;将上述沉淀静置陈化24h;真空抽滤,并用去
离子水洗涤至无cl-为止,将其置于红外线快速干燥器中干燥;研磨,添加20gγ-al2o3,混合均匀,研磨过100目筛得混合物粉末;将混合物粉末浸渍于300g 10%的过硫酸铵溶液后;真空抽滤,红外烘干,在马弗炉中于650℃下焙烧3小时,即可得固体酸催化剂s2o
82-/zro2/γ-al2o3。
[0028]
作为优选,步骤(1)采用的反应溶剂选自二甲苯、甲苯、丁醇、异丙醇中的一种或二种以上任意组合;步骤(2)采用的反应溶剂选自二甲苯、甲苯、乙醇、异丙醇中的一种或二种以上任意组合;步骤(3)采用的反应溶剂选自dmac、nmp、dmf、dme中的一种或二种以上任意组合;步骤(4)采用的反应溶剂选自甲醇、乙醇、dmf、thf中的一种或二种以上任意组合;步骤(5)采用的反应溶剂选自甲苯、乙腈、dmf、thf中的一种或二种以上任意组合;步骤(6)采用的反应溶剂选自甲苯、二甲苯、氯苯、nmp中的一种或二种以上任意组合。
[0029]
作为优选,步骤(1)具体为:在反应器中依次加入异丙醇、4-甲酰基-3-甲氧基苯甲腈、哌啶、醋酸,反应液温度为30℃,开始分批加入乙酰乙酸脂类,维持温度30℃至回流温度,滴加完毕后,同温搅拌1h;反应完毕后降温至0~30℃,搅拌0.5小时析料充分,过滤,压干后用冰异丙醇洗涤两次,得到产物,产物在55℃干燥,得到产品;所述乙酰乙酸脂类选自乙酰乙酸苄酯、乙酰乙酸甲酯、乙酰乙酸乙酯、乙酰乙酸叔丁酯中的一种;4-甲酰基-3-甲氧基苯甲腈、乙酰乙酸脂类、哌啶、醋酸的摩尔比为1:1.1:0.1:0.1。
[0030]
作为优选,步骤(2)具体为:取甲苯加入反应瓶中,加入化合物1和4-氨基-5-甲基吡啶-2-醇,加入对甲苯磺酸,60℃至回流温度反应10~24小时,反应完毕后降温至5℃搅拌0.5h,抽滤,滤饼用冰异丙醇漂洗,得到黄色固体,烘干得到产品;其中,4-氨基-5-甲基吡啶-2-醇、化合物1、对甲苯磺酸的摩尔比为1:1.05:0.21。
[0031]
作为优选,步骤(3)具体为:在反应瓶中加入化合物2,加入溶剂dmf,加入原甲酸三乙酯,加入硫酸,升温至60~150℃反应2~6h,反应完毕后,降温至室温,滴加水析料,加入甲醇搅拌0.5h,析出黄色固体产品,烘干得到产品;其中,化合物2、原甲酸三乙酯、硫酸的摩尔比为1:1.5:0.1。
[0032]
作为优选,步骤(4)具体为:在反应瓶中加入化合物3,加入溶剂无水thf,加入三甲基硅醇钾,室温~60℃反应1~12h,反应结束后,加入乙酸乙酯分液,水相用浓盐酸调节ph至5~6析料,析出固体,用甲醇重结晶,得到类白色固体,烘干得到类白色产品;其中,化合物3、三甲基硅醇钾的摩尔比为1:2。
[0033]
作为优选,步骤(5)具体为:在反应瓶中加thf,加入化合物4,加入cdi,室温~回流反应1~12h,tlc结束后,加入氨水,回流反应1~20h,反应完毕后,加入水稀释反应液,加入乙酸乙酯萃取反应液,反应液盐水洗涤后,干燥,旋蒸至干,得到白色固体;其中,化合物4、cdi、氨水的摩尔比为1:1.1:2~10。
[0034]
作为优选,步骤(6)具体为:反应瓶中加入溶剂二甲苯,加入化合物5,加入催化剂,室温至回流搅拌1~12h,hplc测得目标构型:非目标构型=84:16,结束转构型;过滤,母液旋干,得到粗品,加入乙醇(还可以选用甲醇、异丙醇或丙醇),加入水,搅拌溶解后,加入d-二苯甲酰酒石酸,加热回流1~5h,降温至30℃,搅拌1h过滤,得到粗品,粗品加入水中,加入10%磷酸钠溶液调节ph至7.5,搅拌0.5h析料,过滤得到粗品后,用乙醇重结晶,得到非奈利酮原料药;其中,化合物5、催化剂的摩尔比为1:0.2。
[0035]
本发明第一步通过采用乙酰乙酸甲酯、乙酰乙酸乙酯、乙酰乙酸叔丁酯等常见脂
类代替3-氧代丁酸2-氰乙基酯,这样原料便宜易得,大大降低合成成本和难度;第二步采用甲苯做溶剂,对甲苯磺酸催化,回流反应就能得到产物,这样避免了高压反应,大大提升了反应的安全性和生产效率;最后一步拆分后,另一构型采用二甲苯作溶剂,加催化剂s2o
82-/zro2/γ-al2o3,高温回流,可以将构型转为需要的构型,这样大大提升了收率。
[0036]
与现有技术相比,本发明具有的有益效果在于:
[0037]
1.本发明避免了高压反应,提高了反应效率和安全性;
[0038]
2.催化剂具有活性高、稳定性好、活性组分分散度高、使用寿命长、可重复使用多次等优点,制备流程短、操作简单、反应容易控制、设备要求简单;
[0039]
3.利用催化剂提高构型比例,高产量得到原料药;
[0040]
4.通过采用乙酰乙酸苄酯,乙酰乙酸甲酯、乙酰乙酸乙酯、乙酰乙酸叔丁酯等常见c1-c7脂类代替3-氧代丁酸2-氰乙基酯,原料易得,降低成本,高效合成非奈利酮。
附图说明
[0041]
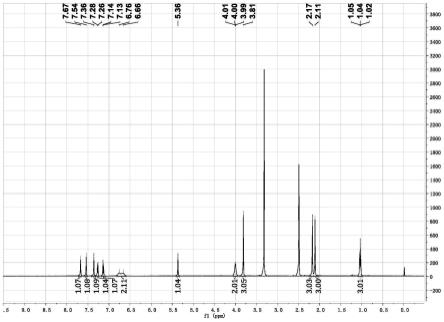
图1是制备得到的非奈利酮核磁谱图。
具体实施方式
[0042]
下面结合具体实施例对发明作进一步说明,但发明的保护范围并不限于此。本领域的普通技术人员可以且应当知晓任何基于本发明实质精神的简单变化或者替换均应属于本发明所要求的保护范围。
[0043]
文中涉及的原料如无特殊说明均购于市场,文中涉及的百分比如无特殊说明为质量百分比。
[0044]
催化剂s2o
82-/zro2/γ-al2o3的制备:
[0045]
称取50g的八水合氧氯化锆,加450g的去离子水配制成质量分数为10%的溶液,在匀速搅拌的状态下,慢慢滴加氨水,至ph到10,停止加氨水此时生成白色沉淀zr(oh)4;将上述沉淀静置陈化24h;真空抽滤,并用去离子水洗涤至无cl-为止,将其置于红外线快速干燥器中干燥;研磨,添加20gγ-al2o3,混合均匀,研磨过100目筛得混合物粉末;将混合物粉末浸渍于300g 10%的过硫酸铵溶液后;真空抽滤,红外烘干,在马弗炉中于650℃下焙烧3小时,即可得固体酸催化剂s2o
82-/zro2/γ-al2o3。
[0046]
实施例1
[0047]
(1)化合物1的合成:
[0048][0049]
在反应器中依次加入150g异丙醇,加入33.8g(0.21mol)4-甲酰基-3-甲氧基苯甲腈,1.86g(0.021mol)哌啶,1.26g(0.021mol)醋酸,反应液温度t=30℃,开始分批加入(0.231mol)乙酰乙酸脂类,维持温度35-40℃,滴加完毕后,同温搅拌1h。hplc检测,开始降温,降温至t=0-3℃搅拌0.5小时析料充分,过滤,压干后用冰异丙醇洗涤两次,得到产物,
5-甲基吡啶-2-醇,加入1.44g(0.0084mol)对甲苯磺酸,升温至回流反应16小时。tlc检测反应完全,出现大量黄色固体,停止反应。降温至t=5℃搅拌0.5h,抽滤,滤饼用少量冰异丙醇漂洗,得到黄色固体,烘干得到黄色固体16.3g,收率92%。
[0055]
(3)化合物3的合成:
[0056][0057]
在反应瓶中加入15.4g(0.035mol)化合物2,加入溶剂80g dmf,加入7.8g(0.053mol)原甲酸三乙酯,加入0.35g(0.0035mol)硫酸,升温至t=120℃反应4h。反应完毕后,降温至室温,慢慢滴加水析料,加入10g甲醇搅拌0.5h,析出黄色固体产品,烘干得到14.7产品,收率90%。
[0058]
(4)化合物4的合成:
[0059][0060]
在反应瓶中加入14.4g(0.0308mol)化合物3,加入溶剂无水thf 80g,加入7.88g(0.0616mol)三甲基硅醇钾,保持t=30℃反应8h,hplc检测合格后,反应结束后,加入80g乙酸乙酯分液,水相用浓盐酸调节ph=5-6析料,析出固体,用20g甲醇重结晶,得到类白色固体,烘干得到类白色产品10.5g,收率90%。
[0061]
(5)化合物5的合成:
[0062][0063]
在反应瓶中加入thf 60g,加入9g(0.0237mol)化合物4,加入4.23g(0.0261mol)cdi,升温至60℃反应8h,tlc结束后,加入6.45g 25%氨水,回流反应12h,tlc检测反应完毕后,加入水稀释反应液,加入乙酸乙酯萃取反应液,反应液盐水洗涤后,干燥,旋蒸至干,得到白色固体7.88g,收率88%。
[0064]
(6)化合物6的合成:
[0065][0066]
反应瓶中加入溶剂二甲苯,加入7g(0.0185mol)化合物5,加入1.4g催化剂s2o
82-/zro2/γ-al2o3,回流搅拌8h。hplc测得目标构型:非目标构型=84:16,结束转构型。过滤,母液旋干,得到粗品,加入15g乙醇,加入5g水,搅拌溶解后,加入5.84g(0.0155mol)d-二苯甲酰酒石酸,加热回流3h,降温至30℃,搅拌1h过滤,得到粗品,粗品加入水中,加入10%磷酸钠溶液调节ph=7.5,搅拌0.5h析料,过滤得到粗品后,用20g乙醇重结晶,得到非奈利酮原料药5.6g,收率80%。制备得到的非奈利酮核磁谱图如图1所示。
[0067]
实施例2
[0068]
(1)化合物1的合成:
[0069]
在反应器中依次加入150g异丙醇,加入33.8g(0.21mol)4-甲酰基-3-甲氧基苯甲腈,1.86g(0.021mol)哌啶,1.26g(0.021mol)醋酸,反应液温度t=30℃,开始分批加入44.3g(0.231mol)乙酰乙酸苄酯,维持温度35-40℃,滴加完毕后,同温搅拌1h。hplc检测,开始降温。降温至t=0-3℃搅拌0.5小时析料充分,过滤,压干后用冰异丙醇洗涤两次,得到产物,产物t=55℃干燥,得到产品64g,收率91%。
[0070]
(2)化合物2的合成:
[0071]
取80g甲苯加入反应瓶中,加入14.07g(0.042mol)化合物1和5g(0.04mol)4-氨基-5-甲基吡啶-2-醇,加入1.44g(0.0084mol)对甲苯磺酸,升温至80℃反应26小时。tlc检测反应完全,出现大量黄色固体,停止反应。降温至t=5℃搅拌0.5h,抽滤,滤饼用少量冰异丙醇漂洗,得到黄色固体,烘干得到黄色固体13.6g,收率77%。
[0072]
(3)化合物3的合成:
[0073]
在反应瓶中加入15.4g(0.035mol)化合物2,加入溶剂80gdmf,加入6.1g(0.042mol)原甲酸三乙酯,加入0.35g(0.0035mol)硫酸,升温至t=110℃反应4h。反应完毕后,降温至室温,慢慢滴加水析料,加入10g甲醇搅拌0.5h,析出黄色固体产品,烘干得到12.6产品,收率76.8%。
[0074]
(4)化合物4的合成:
[0075]
在反应瓶中加入14.4g(0.0308mol)化合物3,加入溶剂无水thf 80g,加入5.91g(0.0462mol)三甲基硅醇钾,保持t=40℃反应8h,hplc检测合格后,反应结束后,加入80g乙酸乙酯分液,水相用浓盐酸调节ph=5-6析料,析出固体,用20g甲醇重结晶,得到类白色固体,烘干得到类白色产品8.6g,收率73.7%。ei-ms m/z:380.4[m h]
。
[0076]
(5)化合物5的合成:
[0077]
在反应瓶中加入thf 60g,加入9g(0.0237mol)化合物4,加入4.23g(0.0261mol)cdi,升温至60℃反应8h,tlc结束后,加入12.8g 25%氨水,回流反应12h,tlc检测反应完毕后,加入水稀释反应液,加入乙酸乙酯萃取反应液,反应液盐水洗涤后,干燥,旋蒸至干,得到白色固体6.9g,收率77%。
[0078]
(6)化合物6的合成:
[0079]
反应瓶中加入溶剂二甲苯,加入7g(0.0185mol)化合物5,加入2.8g催化剂,回流搅拌8h。hplc测得目标构型:非目标构型=84:16,结束转构型。过滤,母液旋干,得到粗品,加入15g乙醇,加入5g水,搅拌溶解后,加入5.84g(0.0155mol)d-二苯甲酰酒石酸,加热回流3h,降温至30℃,搅拌1h过滤,得到粗品,粗品加入水中,加入10%磷酸钠溶液调节ph=7.5,搅拌0.5h析料,过滤得到粗品后,用20g乙醇重结晶,得到非奈利酮原料药5.6g,收率80%。
[0080]
实施例3
[0081]
(1)化合物1的合成:
[0082]
在反应器中依次加入1500g异丙醇,加入338g(2.1mol)4-甲酰基-3-甲氧基苯甲腈,37.2g(0.42mol)哌啶,25.2g(0.42mol)醋酸,反应液温度t=30℃,开始分批加入443g(2.31mol)乙酰乙酸苄酯,维持温度35-40℃,滴加完毕后,同温搅拌1h。hplc检测,开始降温。降温至t=0-3℃搅拌0.5小时析料充分,过滤,压干后用冰异丙醇洗涤两次,得到产物,产物t=55℃干燥,得到产品647.22g,收率92%。ei-ms m/z:336.3[m h]
。
[0083]
(2)化合物2的合成:
[0084]
取500g甲苯加入反应瓶中,加入201g(0.6mol)化合物1和50g(0.4mol)4-氨基-5-甲基吡啶-2-醇,加入14.4g(0.084mol)对甲苯磺酸,升温至回流反应16小时。tlc检测反应完全,出现大量黄色固体,停止反应。降温至t=5℃搅拌0.5h,抽滤,滤饼用少量冰异丙醇漂洗,得到黄色固体,烘干得到黄色固体161.46g,收率90.8%。ei-ms m/z:442.2[m h]
。
[0085]
(3)化合物3的合成:
[0086]
在反应瓶中加入154.3g(0.35mol)化合物2,加入溶剂1000g dmf,加入78g(0.53mol)原甲酸三乙酯,加入3.5g(0.035mol)硫酸,升温至t=130℃反应4h。反应完毕后,降温至室温,慢慢滴加水析料,加入50g甲醇搅拌0.5h,析出黄色固体产品,烘干得到147.7产品,收率90%。ei-ms m/z:470.4[m h]
。
[0087]
(4)化合物4的合成:
[0088]
在反应瓶中加入144.4g(0.308mol)化合物3,加入溶剂无水thf 800g,加入51.2g(0.4mol)三甲基硅醇钾,保持t=30℃反应8h,hplc检测合格后,反应结束后,加入800g乙酸乙酯分液,水相用浓盐酸调节ph=5-6析料,析出固体,用300g甲醇重结晶,得到类白色固体,烘干得到类白色产品95g,收率81.4%。ei-ms m/z:380.3[m h]
。
[0089]
(5)化合物5的合成:
[0090]
在反应瓶中加入thf 600g,加入90g(0.237mol)化合物4,加入57.5g(0.355mol)cdi,升温至60℃反应8h,tlc结束后,加入160g 25%氨水,回流反应12h,tlc检测反应完毕后,加入水稀释反应液,加入乙酸乙酯萃取反应液,反应液盐水洗涤后,干燥,旋蒸至干,得到白色固体78g,收率87%。ei-ms m/z:379.4[m h]
。
[0091]
(6)化合物6的合成:
[0092]
反应瓶中加入溶剂二甲苯,加入70g(0.185mol)化合物5,加入30g催化剂,回流搅拌8h。hplc测得目标构型:非目标构型=84:16,结束转构型。过滤,母液旋干,得到粗品,加入150g乙醇,加入50g水,搅拌溶解后,加入58.4g(0.155mol)d-二苯甲酰酒石酸,加热回流3h,降温至30℃,搅拌1h过滤,得到粗品,粗品加入水中,加入10%磷酸钠溶液调节ph=7.5,搅拌0.5h析料,过滤得到粗品后,用200g乙醇重结晶,得到非奈利酮原料药56g,收率80%。
ei-ms m/z:379.4[m h]
。
[0093]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。