amg510化合物的晶型及其制备方法和用途
技术领域
1.本发明涉及药物化学领域。具体而言,涉及amg510化合物的晶型及其制备方法和用途。
背景技术:
2.kras基因编码蛋白是细胞内信号传导途径中的一种信号传导蛋白,对细胞的生长存活和分化等功能具有重要的影响。当kras基因突变时,不能产生正常的ras蛋白,使细胞内信号传导紊乱,细胞增殖失控而癌变。kras g12c突变通常发生在约13%的肺癌患者、3%的结直肠癌和阑尾癌患者以及1%~3%的其他实体瘤患者中。kras是癌基因ras家族的成员,其突变可能诱导组成性信号转导,导致肿瘤细胞生长、增殖、侵袭和转移。
3.2021年5月28日,fda批准了安进公司的首个kras靶向药lumakras(即sotorasib,曾名为amg510)用于携带kras g12c突变的非小细胞肺癌,结束了这一“最强”致癌突变无药可医的历史。amg510是一种口服的kras g12c小分子抑制剂,靶向致癌性kras取代突变g12c,具有良好的抗肿瘤活性。
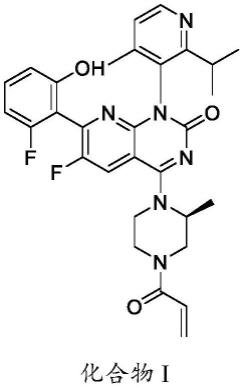
4.amg510化合物的化学名称为6-氟-7-(2-氟-6-羟基苯基)-1-(4-甲基-2-丙烷-2-基吡啶-3-基)-4-[(2s)-2-甲基-4-丙-2-烯酰基哌嗪-1-基]吡啶[2,3-d]嘧啶-2-酮(以下称为“化合物i”),其结构式如下:
[0005][0006]
晶型是化合物固体分子在微观三维结构中长程有序排列而形成晶格的固体形态。药物多晶型现象是指固体药物分子存在两种或两种以上的不同晶型的现象。因为不同的晶型其理化性质不同,固态药物分子的不同晶型可能在体内有不同的溶出、吸收,进而在一定程度上影响药物的临床疗效和安全性,特别是对于难溶性固体药物而言,晶型对生物利用度的影响会更大。因此,药物晶型是固态药物研究及开发过程中的重要一环,也是药物质量控制的重要内容。
[0007]
原研专利us20200369662和wo2020236947中报道了无水晶型i~iii,水合物晶型i,以及thf溶剂合物、mecn溶剂合物、mek溶剂合物、dcm溶剂合物、丙酮溶剂合物、甲醇溶剂
合物、异丙醇溶剂合物和乙醇溶剂合物。该专利文本中描述,无水晶型i是热力学最稳定的晶型,无水晶型i虽然稳定性好,但是存在溶解度低的不足。无水晶型ii和无水晶型iii都会转化为无水晶型i,并且无水晶型ii和iii均具有引湿性,药用开发存在不足。此外,水合物晶型制备过程耗时13天,生产效率很低。其余溶剂合物因含有有机溶剂,毒副作用较大,不可做为药用晶型。
[0008]
综上所述,本领域急需一种具有良好溶解度和稳定性的amg510新晶型,以满足药物的生物利用度、适合产业化开发及各方面综合性能均满足药用开发的新晶型。
[0009]
本技术的发明人意外发現了本发明提供的化合物i的不同晶型,其在理化性质、制剂加工性能及生物利用度等方面具有优势,例如在熔点、溶解度、引湿性、提纯作用、稳定性、黏附性、可压性、流动性、体内外溶出、生物有效性等方面中的至少一方面存在优势,为含化合物i的药物开发提供了更好的选择,具有非常重要的意义。
技术实现要素:
[0010]
本发明的主要目的是提供化合物i的新晶型及其制备方法和用途。
[0011]
根据本发明的目的,本发明提供化合物i的晶型。
[0012]
进一步地,本发明提供化合物i的晶型可以为晶型dciii(以下称为晶型dciii)。
[0013]
一方面,使用cu-ka辐射,所述晶型dciii的x射线粉末衍射图在衍射角2theta值为6.3
°±
0.2
°
,9.0
°±
0.2
°
,14.8
°±
0.2中的1处、或2处、或3处有特征峰。
[0014]
进一步地,使用cu-ka辐射,所述晶型dciii的x射线粉末衍射图在衍射角2theta值还为17.8
°±
0.2
°
,12.7
°±
0.2
°
,16.5
°±
0.2
°
中的1处、或2处、或3处有特征峰。优选地,所述晶型dciii的x射线粉末衍射图在衍射角2theta值还为17.8
°±
0.2
°
,12.7
°±
0.2
°
,16.5
°±
0.2
°
中的3处有特征峰。
[0015]
进一步地,使用cu-ka辐射,所述晶型dciii的x射线粉末衍射图在衍射角2theta值还为12.2
°±
0.2
°
,13.4
°±
0.2
°
,20.3
°±
0.2
°
中的1处、或2处、或3处有特征峰。优选地,所述晶型dciii的x射线粉末衍射图在衍射角2theta值还为12.2
°±
0.2
°
,13.4
°±
0.2
°
,20.3
°±
0.2
°
中的3处有特征峰。
[0016]
另一方面,使用cu-ka辐射,所述晶型dciii的x射线粉末衍射图在衍射角2theta值还为6.3
°±
0.2
°
,9.0
°±
0.2
°
,12.2
°±
0.2
°
,12.7
°±
0.2
°
,13.4
°±
0.2
°
,14.8
°±
0.2
°
,16.5
°±
0.2
°
,17.8
°±
0.2
°
,20.3
°±
0.2
°
,23.2
°±
0.2
°
,27.8
°±
0.2
°
中的1处、或2处、或3处、或4处、或5处、或6处、或7处、或8处、或9处、或10处、或11处具有特征峰。
[0017]
非限制性的,晶型dciii的x-射线粉末衍射图基本如图3所示。
[0018]
非限制性地,晶型dciii在289度附近开始出现吸热峰,差示扫描量热分析图基本如图4所示。
[0019]
非限制性地,晶型dciii是无水晶型。
[0020]
根据本发明的目的,本发明还提供所述晶型dciii的制备方法,所述制备方法包括:
[0021]
方法一:
[0022]
称取一定量的化合物i,加入一定量的有机溶剂或有机溶剂的混合溶剂,搅拌,在一定温度下重结晶,离心分离固体后,可得到晶型dciii。
[0023]
具体地,称取一定量的化合物i加入玻璃瓶中,加入一定量的有机溶剂或有机溶剂的混合溶剂(比如醚类,或酯类,或烷烃类、或腈类、或醇类、或它们二者的混合溶剂体系),并充分震荡,在一定温度下重结晶,离心分离固体后,可得到晶型dciii。
[0024]
进一步地,醚类可以为甲基叔丁基醚;烷烃类可以为正己烷;醇类可以为正丁醇。
[0025]
进一步地,在5℃
±
2℃下搅拌(或者震荡),离心分离固体后,30℃
±
2℃干燥,得到晶型dciii。
[0026]
在一些实施方案中,称取一定量的化合物i,加入一定量的醇类或含有醇类溶剂的混合溶剂,将样品溶清,然后加入烷烃类试剂,在一定温度下搅拌一段时间后,离心分离固体后,可得到晶型dciii。
[0027]
具体地,称取一定量的化合物i加入玻璃瓶中,加入一定量的醇类或含有醇类溶剂的混合溶剂,将样品溶清,然后加入烷烃类试剂,在一定温度下搅拌一段时间后,离心分离固体后,可得到晶型dciii。
[0028]
进一步地,醇类为正丁醇;含有醇类溶剂的混合溶剂为含有正丁醇的甲基叔丁基醚混合溶剂或含有正丁醇的正己烷混合溶剂。
[0029]
进一步地,在5℃
±
2℃下搅拌,离心分离固体后,得到晶型dciii。
[0030]
方法二:
[0031]
称量一定量的化合物i,加入一定量的水形成悬浮液,搅拌一段时间后,分离固体,并将固体加热至高温,收集固体可得晶型dciii。
[0032]
具体地,称量一定量的化合物i加入小瓶中,加入一定量的水形成悬浮液,搅拌一段时间后,分离固体,并将固体加热至高温,收集固体可得晶型dciii。
[0033]
进一步的,所选高温优选190度~235度,更优选220度。
[0034]
本发明提供的晶型dciii具有如下有益效果:
[0035]
1)与现有技术相比,本发明晶型dciii具有更高的溶解度。
[0036]
与现有技术相比,本发明晶型dciii在sgf(模拟胃液),fassif(禁食状态模拟肠液),fessif(进食状态模拟肠液)以及纯水中,均具有更高的溶解度。在1小时,4小时,24小时,本发明晶型dciii的溶解度是现有技术us20200369662a1中报道无水晶型i的2~3倍。更高的溶解度有利于提高药物在人体内的吸收,提高药物的生物利用度,能用更少的载药量达到更好的治疗效果;此外,在保证药品疗效的前提下,降低药物载药量,可降低药物的毒副作用,提高药物使用的安全性,具有重要的临床意义。
[0037]
2)本发明提供的晶型dciii具有良好的稳定性。
[0038]
本发明晶型dciii在25℃/60%rh(相对湿度),40℃/75%rh,60℃/75%rh条件下,分别密闭放置1个月,晶型均保持不变,说明晶型dciii具有良好的物理稳定性,尤其是加速条件40℃/75%rh,以及高温高湿条件60℃/75%rh,放置一个月仍保持晶型稳定,未发生转晶,这进一步说明了晶型dciii即使在高温高湿度条件下,依然具有较好的物理稳定性,这就保证了药物在后续工艺、生产及运输过程中不易发生转晶;此外,晶型dciii在25℃/60%rh(相对湿度)条件下放置前后,化学纯度也未发生变化,纯度均保持在99%以上,说明晶型dciii具有良好的化学稳定性,此外,即使在加速条件40℃/75%rh,以及高温高湿条件60℃/75%rh,化学纯度也依然未发生明显的下降,由此进一步说明了晶型dciii具有良好的化学稳定。良好的物理化学稳定性,保证了药品在后续制剂开发及工艺生产过程,以及药品
生产运输过程中,能够保持质量稳定,确保药物质量及疗效,具有重要的意义。
[0039]
此外,晶型dciii具有良好的机械稳定性。晶型dciii在研磨前后,未发生转晶,且样品结晶度未观察到明显的下降,由此说明晶型dciii具有良好的机械稳定性。良好的机械稳定性可确保样品在后期制剂工艺过程中,不会因为机械研磨、粉碎等外力轻易发生转晶,降低了制剂工艺过程转晶的风险,提高了制剂工艺的可开发性。
[0040]
晶型稳定对药物开发具有重要意义,若发生转晶,将会直接影响药物的溶解度进而影响药品的生物利用度,从而改变药品的疗效。良好的化学稳定性可以确保药品在储存过程中几乎不产生新的杂质或杂质含量几乎不增加,从而确保药品的安全性。良好的机械稳定性也可提高药品在制剂工艺过程中耐受机械力的损伤,降低转晶风险。因此,晶型dciii良好的物理化学稳定性,以及良好的机械稳定性,为后续药物的生产及开发提供了保障,具有较高的产业化开发价值。
[0041]
进一步地,本发明的晶型dciii还具有如下有益效果:
[0042]
1)本发明晶型dciii具有较低的引湿性。
[0043]
根据药典(中国药典2020年版通则9103药物引湿性实验指导原则,实验条件:25
±
1℃,80%相对湿度)方法,考察了本发明晶型dciii的引湿性,结果表明晶型dciii引湿增重0.6%。此外,关于引湿性特征描述与引湿性增重的界定(中国药典2020年版通则9103药物引湿性实验指导原则,实验条件:25
±
1℃,80%相对湿度)原则,晶型dciii增重范畴为:引湿增重小于2.0%但不小于0.2%,属于略有引湿性。该结果表明,晶型dciii具有较低的引湿性。较低的引湿性可以确保样品在后期生产、加工以及储存运输过程中,能够保持较低的引湿增重而不发生潮解现象,从而确保药品质量稳定。
[0044]
2)本发明晶型dciii具有较好的提纯效果,非常适合工业化生产。
[0045]
经过重结晶制备成本发明的晶型dciii后,样品的化学纯度由98.4%提升至99.1%,表明晶型dciii有较好的提纯排杂效果,不仅提高了药物的质量及安全性,也非常适合工业化大规模生产。
[0046]
根据本发明的目的,本发明还提供一种药物组合物,所述药物组合物包含有效治疗量的化合物i的晶型dciii及药学上可接受的载体或辅料。
[0047]
进一步地,本发明提供化合物i的晶型dciii在制备kras g12c抑制剂药物中的用途。
[0048]
更进一步地,本发明提供化合物i的晶型dciii在制备治疗非小细胞肺癌、结直肠癌或阑尾癌药物中的用途。
[0049]
本发明中,所述“搅拌”,采用本领域的常规方法完成,例如磁力搅拌或机械搅拌,搅拌速度为50-1800转/分钟,其中,磁力搅拌优选为300-900转/分钟,机械搅拌优选为100-300转/分钟。
[0050]
所述“分离”,采用本领域的常规方法完成,例如离心或过滤,“离心”的操作为:将欲分离的样品置于离心管中,以10000转/分速率进行离心,至固体全部沉至离心管底部。
[0051]
所述“干燥”可以在室温或更高的温度下进行。干燥温度为室温到约50℃,或者到40℃。干燥时间可以为2~48小时,或者过夜。干燥在通风橱、鼓风烘箱或真空烘箱里进行。
[0052]
本发明中,“晶体”或“多晶型”指被x射线粉末衍射图表征证实的固体。本领域技术人员能够理解,这里所讨论的理化性质可以被表征,其中的实验误差取决于仪器的条件、样
品的准备和样品的纯度,特别是,本领域技术人员公知,x射线粉末衍射图通常会随着仪器条件的不同而有所改变,特別需要指出的是,x射线粉末衍射图中衍射峰的相对强度也可能随着实验条件的变化而变化,所以衍射峰强度的顺序不能作为唯一或决定性因素。事实上,x射线粉末衍射图中衍射峰的相对强度与晶体的择优取向有关,本发明所示的衍射峰强度为说明性而非用于绝对比较。另外,衍射峰位置的实验误差通常在5%或更少,这些位置的误差也应该被考虑进去,通常允许有
±
0.2的误差。另外,由于样品厚度等实验因素的影响,会造成衍射峰角度的整体偏移,通常允许一定的偏移。因而,本领城技术人员可以理解的是,本发明保护晶型的x射线粉末衍射图不必和这里所指的实施例中的x射线粉末衍射图完全一致,任何具有和这些图谱中的特征峰相同或相似的x射线粉末衍射图的晶型均属于本发明的范畴之内。
[0053]
本领域技术人员能够将本发明所列的x射线粉末衍射图和一个未知晶型的x射线粉末衍射图相比较,以证实这两组图反映的是相同还是不同的晶型。
[0054]
在一些实施方案中,本发明的晶型dciii是纯的,基本没有混合任何其他晶型。本发明中“基本没有”当用来指新晶型时指这个晶型含有少于20%(重量)的其他晶型,尤其指少于10%(重量)的其他晶型,更指少于5%(重量)的其他晶型,更指少于1%(重量)的其他晶型。
[0055]
本发明中术语“约”,当用来指可測量的数值时,例如质量、时间、温度等,意味着可围绕具体数值有一定的浮动的范围,该范围可以为
±
10%,
±
5%,
±
1%,
±
0.5%或
±
0.1%。
[0056]
本发明中用“度”来作为温度的单位,应理解为摄氏度。
附图说明
[0057]
图1为根据实施例1a所得晶型dciii的xrpd图;
[0058]
图2为根据实施例1a所得晶型dciii的dsc图;
[0059]
图3为根据实施例1b所得晶型dciii的xrpd图;
[0060]
图4为根据实施例1b所得晶型dciii的dsc图;
[0061]
图5为根据实施例1c所得晶型dciii的xrpd图;
[0062]
图6晶型dciii放置稳定性后xrpd叠图(从上到下依次为:起始晶型dciii,晶型dciii在25℃/60%rh放置一个月,在40℃/75%rh放置一个月,在60℃/75%rh放置一个月);
[0063]
图7晶型dciii研磨前后xrpd叠图(上面为研磨前的xrpd图,下面为研磨后的xrpd图)。
具体实施方式
[0064]
结合以下实施例对本发明做详细说明,所述实施例详细描述本发明的晶型的制备和使用方法。对本领域技术人员显而易见的是,对于材料和方法两者的许多改变可在不脱离本发明范围的情况下实施。
[0065]
本发明中所用到的缩写的解释如下:
[0066]
xrpd:x射线粉末衍射
[0067]
dsc:差示扫描量热
[0068]
采集数据所用的仪器及方法:
[0069]
本发明所述的x射线粉末衍射图在bruker d2 phaser x射线粉末衍射仪上采集。
[0070]
本发明所述的x射线粉末衍射的方法参数如下:
[0071]
x射线光源:cu ka
[0072]
kal(a):1.54060;ka2(a)1.54439
[0073]
ka2/ka1强度比例:0.50
[0074]
电压:30千伏特(kv)
[0075]
电流:10毫安培(ma)
[0076]
扫描范围:自3.0至40.0度
[0077]
本发明所述的差示扫描量热分析(dsc)图在梅特勒dsc3上采集,差示扫描量热分析(dsc)的方法参数如下:
[0078]
扫描速率:10℃/min
[0079]
保护气体:氮气
[0080]
除非特殊说明,以下实施例均在室温条件下操作,所述“室温”不是特定的温度值,是指10-30℃温度范围。
[0081]
根据本发明,作为原料的所述化合物i和/或其盐包括但不限于固体形式(结晶或无定形)、油状、液体形式和溶液。优选地,作为原料的化合物i和/或其盐为固体形式。
[0082]
以下实施例中所使用的化合物i可根据文献所记載的方法制备获得。
[0083]
实施例1:晶型dciii的制备方法
[0084]
实施例1a:
[0085]
称量1g化合物i加入玻璃瓶中,在室温下加入10ml水并充分震荡,放置于磁力搅拌器上搅拌过夜,离心分离固体后,取约10毫克样品,以10度/分钟的速度加热至220度,在220度停留2分钟后取出固体测试xrpd,得到晶型dciii。
[0086]
实施例1a所得晶型dciii的xrpd图如图1所示,xrpd数据如表1所示。
[0087]
实施例1a所得晶型dciii的dsc图如图2所示。
[0088]
表1
[0089]
衍射角2thetad值相对强度%6.3014.0313.238.969.8722.5110.718.2613.0412.207.2628.2712.657.0052.5113.396.6154.2514.226.2323.1814.835.9766.0315.955.5620.4516.495.3757.3817.764.99100.00
18.544.7929.7218.874.7025.4619.854.4740.4420.254.3980.4722.613.9331.4523.153.8443.5723.523.7831.5625.133.5430.3125.683.4731.4426.473.3725.1227.223.2819.3127.843.2137.8628.763.1024.0730.102.9717.6932.112.7911.1032.812.737.6333.442.689.9235.072.569.7336.692.454.11
[0090]
实施例1b:
[0091]
称量1.665g化合物i加入到100ml玻璃瓶中,用移液枪量取13ml正丁醇溶解样品,室温条件下,缓慢滴加20ml甲基叔丁基醚到玻璃瓶中,将玻璃瓶置于5℃环境中搅拌3天,分离固体后30℃干燥。进行xrpd测试,结果显示为本发明所示的晶型dciii。
[0092]
实施例1b所得晶型dciii的xrpd图如图3所示,xrpd数据如表2所示。
[0093]
实施例1b所得晶型dciii的dsc图如图4所示。
[0094]
表2
[0095][0096][0097]
实施例1c:
[0098]
称量15mg化合物i加入到3ml玻璃瓶中,量取0.4ml正丁醇溶解。室温条件下,缓慢滴加0.8ml正己烷到玻璃瓶中,将玻璃瓶置于5℃环境中搅拌1天后,离心分离。收集固体进行xrpd测试,结果显示为本发明所示的晶型dciii。
[0099]
实施例1c所得晶型dciii的xrpd图如图5所示,xrpd数据如表3所示。
[0100]
表3
[0101]
衍射角2thetad值相对强度%6.3413.9416.009.009.8212.61
12.287.2119.3712.736.9544.5213.466.5849.0513.796.4219.4614.266.2110.6714.865.9650.9616.545.3651.7117.715.01100.0018.594.7713.6419.924.4632.2120.234.3957.8620.554.3222.9622.653.9322.4023.163.8421.4725.093.5515.8225.693.4719.6226.513.3614.3227.293.279.3027.893.2027.7328.843.1017.4829.992.986.67
[0102]
实施例2:晶型dciii的动态溶解度
[0103]
进行药物溶解度测试以预测药物体内性能的时候,很重要的一点是尽可能的模拟体内条件,对口服药,用sgf(模拟胃液),fassif(禁食状态模拟肠液),fessif(进食状态模拟肠液)可以模拟体内条件并预测进食的影响,在此类介质中测试的溶解度与人体环境中的溶解度更加接近。
[0104]
分别取us20200369662a1中报道的晶型i(按照公开的方法制备获得)及本发明的晶型dciii各约20mg分别悬浮于1.5ml的sgf,1.5ml的fessif,1.5的ml的fassif及1.5ml的水配置成悬浮液,平衡1小时、4小时和24小时后分别用高效液相色谱法测试溶液中样品的含量(mg/ml),实验结果如下表4、表5所示:
[0105]
表4
[0106][0107]
表5
[0108][0109]
动态溶解度实验结果表明:相较于us20200369662a1报道的无水晶型ⅰ,本发明的晶型dciii在sgf(模拟胃液),fassif(禁食状态模拟肠液),fessif(进食状态模拟肠液)和纯水中,均具有更高的溶解度。
[0110]
实施例3:晶型dciii的稳定性
[0111]
称取本发明制备得到的晶型dciii约5mg,用铝箔袋密封后分别放置在25℃/60%rh,40℃/75%rh,60℃/75%rh条件下,采用hplc和xrpd测定纯度与晶型。实验结果如下表6所示,xrpd叠图如图6所示。
[0112]
表6
[0113]
放置条件放置时间晶型纯度起始——晶型dciii99.44%25℃/60%rh1个月晶型dciii99.45%40℃/75%rh1个月晶型dciii99.49%60℃/75%rh1个月晶型dciii99.50%
[0114]
结果表明:本发明晶型dciii在25℃/60%rh,40℃/75%rh,60℃/75%rh三种条件下,均可保持物理化学稳定至少1个月以上。
[0115]
实施例4:晶型dciii的机械稳定性
[0116]
将晶型dciii置于研钵中,手动研磨5分钟,研磨前后进行xrpd测试。研磨前后xrpd对比如图7所示。结果表明,本发明晶型dciii经过研磨后晶型不变,且结晶度未观察到明显的下降,由此说明晶型dciii具有良好的机械稳定性。
[0117]
实施例5:晶型dciii的引湿性
[0118]
称取本发明晶型dciii约100mg,放置在25
±
1℃,80%相对湿度条件下24小时,记录前后样品的质量。具体结果如下表7所示。
[0119]
关于引湿性特征描述与引湿性增重的界定(中国药典2020年版通则9103药物引湿性实验指导原则,实验条件:25
±
1℃,80%相对湿度):
[0120]
潮解:吸收足量水分形成液体
[0121]
极具引湿性:引湿增重不小于15.0%
[0122]
有引湿性:引湿增重小于15.0%但不小于2.0%
[0123]
略有引湿性:引湿增重小于2.0%但不小于0.2%
[0124]
无或几乎无引湿性:引湿增重小于0.2%。
[0125]
表7
[0126][0127][0128]
结果表明,本发明的晶型dciii略具有引湿性,引湿性较小,由此表明晶型dciii在药品生产及储存过程中不易发生潮解。
[0129]
实施例6:晶型dciii的提纯效果
[0130]
称量30mg纯度为98.4%的样品,加入0.4毫升正丁醇溶清后,缓慢加入2毫升甲基叔丁基醚,然后放置于5度下搅拌过夜,测试固体为晶型dciii,hplc结果显示纯度为99.1%,表明晶型dciii有较好的提纯排杂效果,在工业化生产及放大过程中有较大益处。
[0131]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围,而根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
