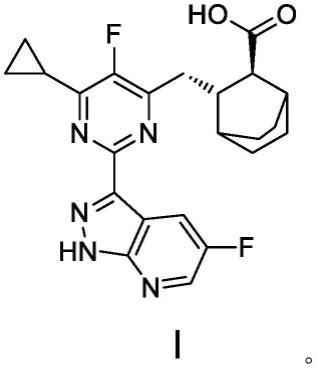
1.本技术属于药物制剂技术领域,具体涉及一种抗流感病毒的药物组合物,尤其是涉及一种嘧啶衍生物类抗流感病毒的干混悬剂。
背景技术:
2.流行性感冒病毒,即流感病毒(influenza virus,ifv),是一种能够导致人和动物患流行感冒的分节状单链反义rna病毒。流感、人患禽流感的大流行已成为全球关注的社会问题和威胁人类健康的重大公共卫生问题。根据世界卫生组织估计,每年流感的季节性流行可导致全球300万例到500万例的重症病例,25万例到50万例的死亡。我国是流感的多发地且多次都是流感首先袭击的地区,每年流感发病数估计可达上千万人。流感和人患禽流感不仅严重危害人体健康甚至危及生命,大流行时也容易引起人群恐慌继而影响社会稳定。流感肆虐也带来很大的经济负担,仅美国,每年成人流感疾病造成的经济损失达800亿美元,成人流感药物市场总额接近100亿美元。
3.目前的流感治疗选择包括接种疫苗和用抗病毒药物进行治疗。流感病毒具有突变率高,病毒间重组现象多的特点,现有的抗流感药物耐药性日趋严重,疫苗作用也常被流感病毒逃逸。
4.化合物1是一种小分子rna聚合酶抑制剂。临床前研究结果表明,化合物i具有很强的体外广谱抗甲型流感病毒活性,对多种甲型流感病毒的抑制能力明显优于同靶点化合物以及神经氨酸酶抑制剂奥司他韦。体内药效试验也较同靶点参考化合物和奥司他韦有更佳的保护动物、降低动物肺部病毒滴度的药效。
[0005][0006]
cn201780054195.x公开了一类抗流感病毒化合物,其中也公开了化合物i在细胞水平抑制流感病毒复制试验中展示出积极效应ec50为0.013nm;在在甲型流感病毒h1n1小鼠感染模型中的药效试验研究中显示在第9天可以实现保护动物体重下降率为4.8%,存活率为100%的效果。
[0007]
然而目前,尚未有关于化合物1制剂的研究,开发出既能有效提高药物的稳定性,又能满足患者使用方便,提高患者顺应性的化合物1的合适药物剂型,有利于化合物1及早
惠及患者,满足临床未被满足的需求。
技术实现要素:
[0008]
本发明的目的在于提供一种化合物i的药物组合物,期望其能够满足临床使用需求,为流感患者提供新的用药选择。
[0009]
具体,本发明提供了一种化合物i的药物组合物,其包含治疗和/或预防有效量的化合物i,以及一种或多种药学可接受的赋形剂。
[0010]
进一步的,本发明提供了一种化合物i的干混悬剂,其中,包含如下重量百分比的组分:活性成分化合物i1%~10%,填充剂70%~90%,矫味剂1%~3%,优选地,化合物i的含量为4%~8%,更优选地,化合物i的含量为4%~6%,
[0011][0012]
进一步的,所述化合物i的干混悬剂,还包括稳定剂4%~7%和/或遮光剂0.5%~2.0%,。优选地所述稳定剂的含量为5%~7%,更优选地,所述稳定剂的含量为6%~7%;
[0013]
进一步的,所述稳定剂为柠檬酸二氢钠,所述遮光剂为二氧化钛。
[0014]
进一步的,所述的干混悬剂,还包括助悬剂1%~2.5%,甜味剂0.1%~0.3%,防腐剂0.1%~0.4%,优选地,所述防腐剂的含量为0.2%~0.3%。
[0015]
优选地,所述填充剂为选自山梨醇、甘露醇、麦芽糖醇或果糖;所述助悬剂为羧甲基纤维素钠或者黄原胶,所述防腐剂为苯甲酸钠,所述矫味剂为橙子味香精,所述甜味剂为糖精钠或阿斯巴甜。
[0016]
在本发明的另一个更优选地实施例中,提供了一种包含化合物i的干混悬剂,包含如下重量百分比的组分:化合物i 3~8%,柠檬酸二氢钠5%~7%,苯甲酸钠0.1%~0.3%,山梨醇80%~90%,黄原胶1%~2.5%,二氧化钛0.5%~2.0%,橙子味香精1%~3%,糖精钠0.1%~0.3%。
[0017]
进一步优选地,所述包含化合物i的干混悬剂,包含如下重量百分比的组分:化合物i 4~8%,柠檬酸二氢钠6%~7%,苯甲酸钠0.2%~0.3%,山梨醇75%~90%,黄原胶1%~2%,二氧化钛1.0%~2.0%,橙子味香精1%~2%,糖精钠0.1%~0.2%。
[0018]
在本发明的另一方面,还提供了所述包含化合物i的口服混悬剂的制备方法,包括以下步骤:
[0019]
(1)预处理:将处方量的原辅料分别进行粉碎后,过80目筛;
[0020]
(2)制备含药颗粒:将处方量的化合物i、30%~45%(w/w)的填充剂、任选的助悬
剂混合均匀,得到含药颗粒;
[0021]
(3)制备空白颗粒:将剩余量填充剂、矫味剂、以及任选的其他辅料包括遮光剂、稳定剂、甜味剂混合均匀,得到空白颗粒;
[0022]
(4)干法制粒:按等量递增法将含药颗粒,空白颗粒和防腐剂混合均匀,干法制粒后过18目筛整粒,分装,即得含化合物i的干混悬剂。
[0023]
在本发明中,优选地,所述口服干混悬剂中化合物i的粒径d(v,0.9)≤50μm,更优选粒径d(v,0.9)≤30μm。
[0024]
根据本发明,粒径d(v,0.9)具有如下的含义,即当d(v,0.9)=50μm时,表示在所描述的整个颗粒群体中,有占总体积90%的粒子的粒径小于等于50μm。
[0025]
本发明所述的药物组合物中化合物i可以以化合物i的可药用盐、或化合物i的水合物的形式存在,也可以以化合物i的游离酸形式存在,所述的药物组合物中化合物i的含量均是指折算成化合物i的游离酸形式后的含量。
[0026]
cn111819177a和cn109790159a公开了化合物i的药学可接受盐,水合物,以及游离形式的制备方法,通过引用结合到本发明中。
[0027]
采用本发明方法制备的口服干混悬剂稳定性好,在高温高湿以长期稳定性试验研究中证明有关杂质出现少,并且本发明制备的口服干混悬剂外观良好,复溶后具有良好的溶出行为,生产简单,剂量方便调整,不仅适合成年人而且特别适合儿童、老年人和吞咽困难病人应用,生物利用度高能够满足儿童、老年人等特殊群里的用药需求和用药依从性。
[0028]
本发明提供的制备方法无粘连,原辅料损失小,有利于改善操作环境和降低生产成本。
具体实施方式
[0029]
需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。
[0030]
实施例1、化合物i的口服干混悬剂的制备
[0031]
处方含量如下所示:
[0032]
[0033][0034]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、33.36%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i的口服干混悬剂,化合物i含量为3mg/100mg。
[0035]
其中化合物i粒径d(v,90)=30μm。
[0036]
实施例2、化合物i的口服干混悬剂的制备
[0037]
处方:
[0038]
[0039][0040]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、33.40%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,即得含化合物i的干混悬剂,化合物i含量为5mg/100mg。
[0041]
其中化合物i粒径d(v,90)=35μm。
[0042]
实施例3、化合物i的口服干混悬剂的制备
[0043]
处方含量如下所示:
[0044]
[0045][0046]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、33.36%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i的口服干混悬剂,化合物i含量为3mg/100mg。
[0047]
其中化合物i粒径d(v,90)=30μm。
[0048]
实施例4、化合物i的口服干混悬剂的制备
[0049]
处方如下所示:
[0050]
[0051][0052]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、32.54%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i的口服干混悬剂,化合物i含量为6mg/100mg。
[0053]
其中化合物i粒径d(v,90)=35μm。
[0054]
实施例5、化合物i的口服干混悬剂的制备
[0055]
处方含量如下所示:
[0056]
[0057][0058]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、34.29%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i的口服干混悬剂,化合物i含量为10mg/100mg。
[0059]
其中化合物i粒径d(v,90)=29μm。
[0060]
实施例6、化合物i的口服干混悬剂的制备
[0061]
处方含量如下所示:
[0062]
[0063][0064]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、39.30%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i的口服干混悬剂,化合物i含量为4mg/100mg。
[0065]
其中化合物i粒径d(v,90)=28μm。
[0066]
实施例7、化合物i一水合物的口服干混悬剂的制备
[0067]
处方含量如下所示:
[0068]
[0069][0070]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、39.30%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i一水合物的口服干混悬剂。
[0071]
其中化合物i一水合物粒径d(v,90)=40μm。
[0072]
其中化合物i一水合物的结构如下所示:
[0073][0074]
所示化合物i一水合物按照wo2019170067公开的方法制备得到。
[0075]
实施例8、包含化合物i盐酸盐的口服干混悬剂的制备
[0076]
处方含量如下所示:
[0077][0078]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i盐酸盐、30.60%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i盐酸盐的口服干混悬剂。
[0079]
其中化合物i盐酸盐粒径d(v,90)=35μm。
[0080]
其中化合物i盐酸盐的结构如下式(ii-1)所示:
[0081][0082]
所述化合物i盐酸盐按照wo2019170067公开的方法制备得到。
[0083]
实施例9、包含化合物i钠盐的口服干混悬剂的制备
[0084]
处方含量如下所示:
[0085]
[0086][0087]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i钠盐、44.91%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,得到含化合物i钠盐的口服干混悬剂。
[0088]
其中化合物i钠盐粒径d(v,90)=32μm。
[0089]
其中化合物i钠盐的结构如下式(iii-1)所示:
[0090][0091]
所述化合物i钠盐按照wo2019170067公开的方法制备得到。
[0092]
实施例10、化合物i的口服干混悬剂的制备
[0093]
处方:
[0094]
成分单剂量(mg)百分比(%)化合物i25.005.00山梨醇418.5083.70黄原胶7.51.50二氧化钛7.51.50柠檬酸二氢钠32.56.50橙子味香精7.251.45糖精钠0.50.10苯甲酸钠1.250.25
合计500100.00
[0095]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、山梨醇、黄原胶、二氧化钛、柠檬酸二氢钠、橙子味香精、糖精钠和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,即得含化合物i的干混悬剂,化合物i含量为5mg/100mg。
[0096]
实施例11、化合物i的口服干混悬剂的制备
[0097]
处方:
[0098][0099][0100]
制备方法:首先,将处方量的原辅料分别进行粉碎后,过80目筛,备用;然后将处方量的化合物i、39.4%(w/w)的山梨醇、黄原胶混合均匀,得到含药颗粒;再将剩余量山梨醇、橙子味香精、阿巴斯甜混合均匀,得到空白颗粒;按等量递增法将含药颗粒,空白颗粒和苯甲酸钠混合均匀,过60目筛;干法制粒后过18目筛整粒,分装,即得含化合物i的干混悬剂,化合物i含量为7mg/100mg。
[0101]
其中化合物i粒径d(v,90)=31μm。
[0102]
实施例12:稳定性研究
[0103]
加速试验条件:将本发明实施例1~11制备的干混悬剂分别放于温度40
±
2℃,相对湿度为75%
±
5%的条件下进行加速试验,放置6个月,分别抽取0,1,3,6月时化合物i干混悬剂进行有关物质、含量的考察。
[0104]
考察结果如下表1所示:
[0105]
表1、加速试验考察结果
[0106]
[0107][0108]
将本发明实施例1~7以及实施例10~11制备的干混悬剂在温度为60
±
2℃,相对湿度为75%
±
5%的条件下,放置10天,采用高效液相色谱法检测化合物i杂质降解情况,所得结果如下表2所示:
[0109]
表2:有关杂质总杂变化情况
[0110][0111]
加速试验效果显示,采用本发明方法制备化合物i或其药学可接受盐的干混悬剂稳定性好,在温度为40
±
2℃,相对湿度为75%
±
5%的条件下,放置6个月单杂含量均没有超过0.1%,化合物i的总杂质及含量变化较小,基本无降解杂质增加。
[0112]
长期试验:去本发明实施例2~3以及10~11制备的化合物i或其可药用盐的干混悬剂放于25
±
2℃,湿度60%
±
10%的条件下进行长期试验,分别抽取0,3,6,9,12月时化合物i或其可药用盐的干混悬剂进行有关物质、含量、溶出度的考察,考察结果如表3所示。
[0113]
表3:长期试验考察结果
[0114]
[0115][0116]
从长期稳定性试验考察结果可以看出,本发明提供的化合物i干混悬剂在室温放置稳定性好,放置12个月含量及有关物质基本没有变化。
[0117]
实施例13溶出性研究
[0118]
根据采用ph1.0作为溶出介质,转速为25rpm,对实施例1~11制备的干混悬剂进行测定溶出曲线。所得结果见表4。
[0119]
表4溶出百分比
[0120][0121][0122]
从以上结果可以看出,本发明方法制备的化合物i干混悬剂3min内溶出度查过75%,30min中内溶出率均能到达100%,具有良好的溶出效果。
[0123]
以上述依据本技术的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项申请技术思想的范围内,进行多样的变更以及修改。本项申请的技术
性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。