1.本发明涉及晶型药物分子技术领域,特别涉及氯唑沙宗晶型技术领域,具体指一种氯唑沙宗-2-吲哚甲酸共晶、其制备方法和应用。
背景技术:
2.药物共晶是指在氢键或其它非共价键作用下药物活性成分与其他物质按一定的化学计量比结合而成的晶体。以氢键为基础的分子网状链接方式是共晶形成的基础。共晶的结合是在中性状态下,依靠的是非离子键结合,当药物的活性成分是中性分子,药物不能通过制备多晶型、无定型,或是制备溶剂化合物和盐来改善药物性质时,共晶是个非常好的选择。药物共晶很多时候不仅不破坏药物的活性,而且能改善药物的熔点、溶解度、稳定性、溶出率以及生物利用度等理化性质。
3.氯唑沙宗(chlorzoxazone),化学名:5-氯-2-苯并噁唑啉酮,英文名:5-chloro-2-benzoxazolinone。cas号:95-25-0,其结构式如下所示:
[0004][0005]
氯唑沙宗是一种口服强效肌肉松弛剂,由美国mcneil公司研制,并于二十世纪六十年代中期上市,临床用于腰背痛、神经痛、风湿性关节炎、急慢性软组织(肌肉、韧带)扭伤、挫伤、运动后肌肉酸痛、中枢神经病变引起的肌肉痉挛及慢性筋膜炎,总有效率高达98.59%。另外对中枢神经病变引起的肌肉痉挛以及慢性筋膜炎、儿童智力发育不良有一定疗效,是目前临床应用最广泛的用药之一。
[0006]
上市氯唑沙宗一般会与对乙酰氨基酚形成复方制剂,复方工艺较为复杂,甚至部分患者对乙酰氨基酚出现过敏现象。鉴于以上不足,本发明提供了氯唑沙宗-2-吲哚甲酸共晶的形式,从而更高效的发挥氯唑沙宗的药用价值。
技术实现要素:
[0007]
本发明提供一种简单且易于操作的制备高纯度氯唑沙宗-2-吲哚甲酸共晶晶型的方法,为氯唑沙宗在药物协同治疗方面的应用提供更好的依据,从而更高效的发挥氯唑沙宗的药用价值。
[0008]
本发明的一个目的是提供一种氯唑沙宗-2-吲哚甲酸共晶;
[0009]
具体技术内容如下:
[0010]
一种氯唑沙宗-2-吲哚甲酸共晶,使用cu-kα辐射,以2θ表示的x射线衍射谱图在5.82
±
0.2
°
,12.85
±
0.2
°
,14.37
±
0.2
°
,17.87
±
0.2
°
,26.98
±
0.2
°
,27.80
±
0.2
°
有特征峰。
[0011]
优选地,所述的氯唑沙宗-2-吲哚甲酸共晶,使用cu-kα辐射,以2θ表示的x射线衍射谱图在5.82
±
0.2
°
,12.85
±
0.2
°
,13.53
±
0.2
°
,14.37
±
0.2
°
,17.47
±
0.2
°
,17.87
±
0.2
°
,19.65
±
0.2
°
,24.91
±
0.2
°
,26.27
±
0.2
°
,26.98
±
0.2
°
,27.80
±
0.2
°
,29.19
±
0.2
°
,29.60
±
0.2
°
,40.12
±
0.2
°
,处有特征峰。
[0012]
优选地,所述的氯唑沙宗-2-吲哚甲酸共晶,使用cu-kα辐射,其特征峰符合图1及表2所示的x射线粉末衍射图谱及检测数据。
[0013]
优选地,所述的氯唑沙宗-2-吲哚甲酸共晶经差示扫描热分析(dsc)检测出现一个吸热峰,温度范围为156.76~193.43℃,吸热峰的峰值在183.88℃,对应为氯唑沙宗-2-吲哚甲酸分解吸热峰。
[0014]
本发明第二方面提供一种制备所述氯唑沙宗-2-吲哚甲酸共晶的方法,该方法包括以下步骤:
[0015]
将氯唑沙宗和2-吲哚甲酸溶于有机溶剂a中,加热溶解,溶液澄清后,降温析晶,过滤干燥得氯唑沙宗-2-吲哚甲酸共晶体。
[0016]
所述的有机溶剂a选自甲醇、乙醇、丙酮、异丙醇的一种或者至少两种以上的混合溶剂。
[0017]
优选地,所述有机溶剂a选甲醇、乙醇的一种或两种。
[0018]
所述氯唑沙宗与2-吲哚甲酸的摩尔比为1:0.87~1.22;优选地,氯唑沙宗与2-吲哚甲酸的摩尔比为1:0.92~1.11。
[0019]
所述体系中2-吲哚甲酸和有机溶剂a的质量体积比为5~13:1,其中质量以mg计,体积以ml计。
[0020]
所述的溶解加热的温度为40~55℃。
[0021]
所述的降温析晶温度为0~30℃,优选地,降温析晶温度为5~20℃。
[0022]
所述的析晶时间为4~6小时。
[0023]
所述干燥温度为45~70℃,干燥时间为8~12小时。
[0024]
优选地,所述制备方法包括以下步骤:
[0025]
将氯唑沙宗与2-吲哚甲酸于有机溶剂a中,40~55℃加热溶解,搅拌回流反应1~2小时,降温至5~20℃析晶4~6小时,过滤,洗涤滤饼,干燥得氯唑沙宗-2-吲哚甲酸共晶。
[0026]
所述洗涤滤饼的溶剂选自丙酮、甲醇、乙醇和乙腈中的一种。
[0027]
本发明第三方面提供一种药物组合物,包括上述制备的氯唑沙宗-2-吲哚甲酸共晶,并含有可联合使用的其它活性成分和/或制剂学上可接受的辅料组分。
[0028]
优选的,所述的其它组分包括可联合使用的其它活性成分、赋形剂、填充剂等。
[0029]
优选地,所述药物组合物可使用标准和常规的技术,制成喷雾剂、片剂、胶囊剂、粉针剂、液体注射剂等。
[0030]
本技术的第四方面提供一种氯唑沙宗-2-吲哚甲酸共晶作为活性成分制备镇痛药中的应用。
[0031]
晶体结构的确认
[0032]
x射线晶体数据在日本理学xtalab synergy型号仪器上收集,测试温度293(2)k,用cuka辐射,以ω扫描方式收集数据并进行lp校正。用直接法解析结构,差值傅里叶法找出全部非氢原子,所有碳及氮上的氢原子采用理论加氢得到,采用最小二乘法对结构进行精修。
[0033]
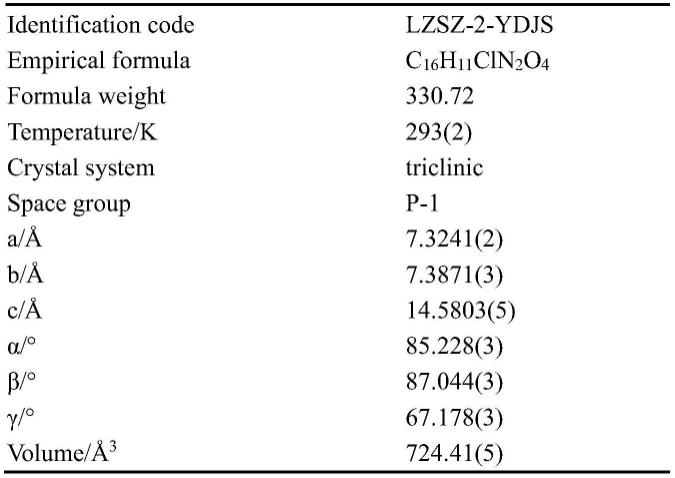
测试及解析本发明制备的氯唑沙宗-2-吲哚甲酸共晶,其晶体学参数:三斜晶系,
手性空间群为p-1;晶胞参数为:α=85.228(3)
°
,β=87.044(3)
°
,γ=67.178(3)
°
,晶胞体积分子式:c
16h11cl
n2o4,分子量:330.72。本发明的氯唑沙宗-2-吲哚甲酸共晶的结构解析ortep图表明,该晶体存在一分子氯唑沙宗,一分子2-吲哚甲酸,如附图3所示。本发明的氯唑沙宗-2-吲哚甲酸共晶的堆积图,如附图2所示。
[0034]
表1氯唑沙宗-2-吲哚甲酸共晶的主要晶体学数据
[0035][0036][0037]
本发明所述氯唑沙宗-2-吲哚甲酸共晶测试中x-射线粉末衍射测试仪器及测试条件为:panalytical empyrean x-射线粉末衍射仪;光源cu靶,平板样品台,入射光路:bbhd,衍射光路:pixcel,电压45kv,电流40ma,发散狭缝为1/4
°
,防散射狭缝为1
°
,索拉狭缝为
0.04rad,每步计数时间0.5s,扫描范围3~50
°
。
[0038]
依据晶体学数据,其对应的x射线粉末衍射图(cu-kα)中特征峰详见附图1及2。
[0039]
表2氯唑沙宗-2-吲哚甲酸共晶主要的pxrd峰
[0040][0041][0042]
实施例中所制备的所有样品都具有上述相同的晶体学参数及x射线粉末衍射谱图。
[0043]
本发明中tga/dsc热分析测试仪及测试条件:tga/dsc热分析仪:mettler toledo tga/dsc3 ;动态温度段:30~300℃;加热速率:10℃/min;程序段气体n2;气体流量:50ml/min;坩埚:铝坩埚40μl。
[0044]
本发明所述方法制备的氯唑沙宗-2-吲哚甲酸共晶,其差示扫描量热曲线(dsc)的结果如图4所示,氯唑沙宗-2-吲哚甲酸共晶经差示扫描热分析(dsc)检测出现一个吸热峰,温度范围为156.76~193.43℃,吸热峰的峰值在183.88℃,对应为氯唑沙宗-2-吲哚甲酸分解吸热峰。其热重分析(tga)只存在一个失重台阶,能够与dsc很好的对应。所述氯唑沙宗-2-吲哚甲酸存在如图4所示的dsc/tga图谱。
[0045]
本发明提供的制备氯唑沙宗-2-吲哚甲酸共晶的方法操作简便,制备的晶体纯度高,本发明提供的氯唑沙宗-2-吲哚甲酸共晶在固态下具有较好的化学稳定性,并且具有较好的溶解度。
附图说明
[0046]
图1:氯唑沙宗-2-吲哚甲酸共晶的x射线粉末衍射图谱。
[0047]
图2:氯唑沙宗-2-吲哚甲酸共晶的堆积图。
[0048]
图3:氯唑沙宗-2-吲哚甲酸共晶的ortep图。
[0049]
图4:氯唑沙宗-2-吲哚甲酸共晶的差示扫描量热曲线(dsc)图。
具体实施方式
[0050]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0051]
实施例1
[0052]
将100.0mg氯唑沙宗,95.13mg2-吲哚甲酸加入到8ml甲醇中,加热到50℃搅拌溶解,回流反应1小时,缓慢降温至10~15℃后,控温静置析晶5小时,过滤,用丙酮洗涤滤饼,55℃下真空干燥10h得氯唑沙宗-2-吲哚甲酸,收率97.81%,纯度99.98%。
[0053]
实施例2
[0054]
将99.8mg氯唑沙宗,99.59mg 2-吲哚甲酸加入到15ml乙醇中,加热到45℃搅拌溶解,回流反应1小时,缓慢降温至5~10℃后,控温静置析晶5小时,过滤,用乙醇洗涤滤饼,45℃下真空干燥8h得氯唑沙宗-2-吲哚甲酸共晶,收率97.27%,纯度99.97%。
[0055]
实施例3
[0056]
将169.57mg氯唑沙宗,158.45mg 2-吲哚甲酸加入到18ml混合溶剂(9ml甲醇和9ml丙酮)中,加热到52℃搅拌溶解,回流反应2小时,缓慢降温至15~20℃后,控温静置析晶4小时,过滤,用乙腈洗涤滤饼,65℃下真空干燥11h得氯唑沙宗-2-吲哚甲酸共晶,收率95.76%,纯度99.96%。
[0057]
实施例4
[0058]
将110.5mg氯唑沙宗,125.70mg 2-吲哚甲酸加入到12ml异丙醇中,加热到55℃搅拌溶解,回流反应1小时,缓慢降温至8~15℃后,控温静置析晶6小时,过滤,用甲醇洗涤滤饼,60℃下真空干燥9h得氯唑沙宗-2-吲哚甲酸共晶,收率95.43%,纯度99.95%。
[0059]
实施例5
[0060]
将80.9mg氯唑沙宗,61.5mg 2-吲哚甲酸加入到4ml丙酮中,加热到35℃搅拌溶解,回流反应1小时,缓慢降温至0~5℃后,控温静置析晶3小时,过滤,用甲醇洗涤滤饼,40℃下真空干燥7h得氯唑沙宗-2-吲哚甲酸共晶,收率78.19%,纯度99.92%。
[0061]
实施例6
[0062]
将66.2mg氯唑沙宗,81.79mg 2-吲哚甲酸加入到21ml混合溶剂(11ml甲醇和10ml乙醇)中,加热到50℃搅拌溶解,回流反应2小时,缓慢降温至20~30℃后,控温静置析晶7小时,过滤,用乙腈洗涤滤饼,75℃下真空干燥14h得氯唑沙宗-2-吲哚甲酸共晶,收率91.42%,纯度99.90%。
[0063]
稳定性试验
[0064]
将本发明实施例1~6中制备的氯唑沙宗-2-吲哚甲酸共晶进行加速试验,置40
±
2℃;rh75
±
5%的恒温恒湿培养箱6个月,分别于0、1、2、3、6个月末取样检验外观、有关物质、
含量及干燥失重。结果见表3。
[0065]
表3氯唑沙宗-2-吲哚甲酸共晶的加速试验结果
[0066][0067]
[0068]
经加速试验表明本发明的氯唑沙宗-2-吲哚甲酸共晶物理化性质较稳定,纯度未见明显降低,杂质含量增加较少。
[0069]
溶解度试验
[0070]
具体的溶解度试验参照中国药典2015分别精密称取实施例中1~6的氯唑沙宗-2-吲哚甲酸共晶过量,置于小西林瓶中,分别加入dmso、ph1.0的盐酸溶液、水,配制成氯唑沙宗饱和溶液,摇匀溶解,过滤。照紫外-可见分光度法(通则0401),在270nm的波长处测定吸光度来计算其溶解度。试验结果见表4。
[0071]
表4氯唑沙宗-2-吲哚甲酸共晶的溶解度
[0072][0073]
经试验得知,本发明方案制备的所有氯唑沙宗-2-吲哚甲酸共晶均可达到相近的溶解度效果,具有较好的溶解度。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。