1.本发明涉及光电材料技术领域,尤其涉及一种过渡金属离子掺杂的钙钛矿量子点材料及其制备方法。
背景技术:
2.化石能源的大规模使用为人类生活提供了巨大的便捷,但同时也带来了环境污染、气候变化等诸多挑战。另一方面,因化石能源不可持续性引起的能源短缺问题也时刻威胁着人类数百年来所建立的工业文明的基石。面对日益恶化的环境和化石能源短缺的问题,发展清洁可持续的新能源供应体系是人类通往未来的必由之路。太阳能作为一种全球分布广泛的能源形式,具有清洁无污染、可再生等优势,是人类可大规模利用并具备现实意义的新能源形式之一,而通过光伏效应将太阳能转变为电能是太阳能利用最为广泛和重要的方式。
3.太阳能电池技术目前已经发展到第三代,钙钛矿太阳能电池作为第三代光伏技术的杰出代表,因其高效率、低成本、可溶液制备等优势,在短短的十余年时间里,得到了飞速的发展。其光电转换效率已经达到25.7%,几乎追平了单晶硅太阳能电池26.1%的记录效率,并具有进一步发展的潜力。但是,目前高效率钙钛矿太阳能电池都是基于有机-无机杂化钙钛矿半导体材料(如mapbi3、fapbi3),由于有机阳离子易挥发易吸水的特性,导致有机-无机杂化钙钛矿材料的湿热稳定性较差,严重制约了其产业化发展。使用无机阳离子cs
替换有机-无机杂化钙钛矿材料中的有机阳离子可以显著提高钙钛矿材料的湿热稳定性,因而基于cs阳离子的全无机钙钛矿材料是高稳定性钙钛矿太阳能电池的重要发展方向。但是由于cs的离子半径较小,难以支撑[pbi6]
4-构成的正八面体空间结构,在室温或湿度环境下[pbi6]
4-正八面体结构容易发生扭曲和倾斜,导致全无机钙钛矿材料cspbi3发生相变,转变为不具备光学活性的非钙钛矿结构。
[0004]
通过元素掺杂,使用离子半径较小的金属离子部分替换cspbi3中的pb离子,从而形成尺寸更小的[bi6]
4-(b为掺杂元素),可以有效改善因cs离子半径较小引起的离子半径不匹配的问题,提高全无机钙钛矿材料的晶体结构稳定性。研究人员通过zn、bi、eu等元素的掺杂,在一定程度上提高了cspbi3材料的热稳定性,但是其湿度稳定性提升仍然十分有限,为真正实现全无机钙钛矿材料的大规模使用和产业化发展,其湿热稳定性仍然需要进一步提高。
[0005]
鉴于此,特提出本发明。
技术实现要素:
[0006]
为了提供一种高稳定性钙钛矿量子点材料,本发明对提高其稳定性的方法进行了探究,以解决全无机钙钛矿量子点材料晶体结构不稳定的问题。
[0007]
具体而言,现有研究表明,由于离子半径较小的cs
难以支撑由pb
2
和i-构成的正八面体空间结构,导致[pbi6]
4-三维空间结构发生扭转和倾斜,这是全无机钙钛矿材料晶体
结构不稳定的根本原因。
[0008]
鉴于此,本发明首先提出通过掺杂较pb小的过渡金属离子,抑制八面体的扭转与倾斜,从而达到稳定晶体结构的目的。
[0009]
具体地,本发明提供过渡金属卤化物在钙钛矿量子点材料中的应用,所述过渡金属卤化物(即掺杂剂)的化学式为bxy;
[0010]
其中,b代表ti、v、cr、mn、fe、co、ni、cu或zn;x代表cl、br或i;y=b的价态/x的价态。
[0011]
本发明从最本质的晶体结构出发,通过引入离子半径较小的过渡金属离子部分替换全无机钙钛矿量子点材料中的pb元素,从而减小正八面体的尺寸,改善钙钛矿晶格中离子半径不匹配的问题,提高全无机钙钛矿量子点材料的晶体结构稳定性。
[0012]
进一步地,当选用如上过渡金属卤化物时,其中的过渡金属离子的离子半径与钙钛矿晶格更匹配,可进一步提高钙钛矿量子点材料的稳定性。
[0013]
具体地,本发明优选过渡金属离子为tivcrmnfeconicucuzn所述过渡金属离子半径均小于pb可以有效改善钙钛矿晶体结构应力状态,提高钙钛矿结构稳定性。
[0014]
作为优选,所述过渡金属卤化物为碘化镍、碘化钴或碘化钒;选用上述过渡金属卤化物作为掺杂剂,可更进一步地提高钙钛矿量子点材料的稳定性。
[0015]
本发明还提供一种掺杂有过渡金属卤化物的钙钛矿量子点材料,其中,所述过渡金属卤化物同上述;
[0016]
所述钙钛矿量子点材料的化学式为cspbx3;其中,x代表cl、br或i。
[0017]
作为优选,所述掺杂有过渡金属卤化物的钙钛矿量子点材料的晶粒尺寸为5-30nm。
[0018]
本发明同时提供以上所述的掺杂有过渡金属卤化物的钙钛矿量子点材料的制备方法,包括如下步骤:
[0019]
s1、将碳酸铯、油酸和1-十八烯混合后干燥,而后加热至碳酸铯完全溶解,得到油酸铯前驱体溶液;
[0020]
将过渡金属卤化物、卤化铅、1-十八烯和助溶剂混合后干燥,而后加入干燥的油胺和油酸,待溶液澄清透明,加热至120-200℃,得卤化铅和过渡金属卤化物的混合溶液;
[0021]
s2、将所述油酸铯前驱体溶液与所述混合溶液快速混合,反应5s-5mins。
[0022]
本发明还发现,通过上述方式,可实现过渡金属离子的有效掺杂;其中,将混合溶液预先加热至反应温度(即120-200℃)后再与油酸铯前驱体溶液混合,可有效控制晶粒尺寸和掺杂比例;一般地,在上述温度范围内,反应温度越高,晶粒尺寸越大,实际掺杂比例越高。
[0023]
作为优选,s1步骤的油酸铯前驱体溶液中,1-十八烯为溶剂,碳酸铯的浓度为10-30mg/ml,油酸的浓度为0.05-0.1ml/ml。
[0024]
作为优选,s1步骤的油酸铯前驱体溶液中,所述干燥在110-130℃下进行0.8-1.2h。
[0025]
作为优选,s1步骤的油酸铯前驱体溶液中,所述加热在惰性气氛下进行;所述加热的温度为140-160℃。
[0026]
作为优选,s1步骤的混合溶液中,1-十八烯为溶剂,卤化铅的浓度为0.02-0.06mmol/ml,助溶剂的浓度为0.1-0.4ml/ml,油胺的浓度为0.1-0.4ml/ml,油酸的浓度为0.1-0.4ml/ml。
[0027]
作为优选,s1步骤的混合溶液中,所述干燥在110-130℃的真空环境下进行0.8-1.2h。
[0028]
作为优选,s1步骤的混合溶液中,所述加热在惰性气氛下进行。
[0029]
作为优选,s1步骤的混合溶液中,以所述卤化铅的物质的量为基准,所述过渡金属卤化物的添加量为0.1-400%;
[0030]
进一步地,以所述卤化铅的物质的量为基准,所述过渡金属卤化物的添加量为100-300%;
[0031]
本发明还发现,通过控制过渡金属卤化物与卤化铅的摩尔比,可更加精准的控制晶粒尺寸和掺杂比例;一般地,在上述配比范围内,过渡金属卤化物与卤化铅的比值越高,晶粒尺寸越小,实际掺杂量越高。
[0032]
作为优选,所述卤化铅为碘化铅。
[0033]
作为优选,所述助溶剂为三正辛基膦。
[0034]
作为优选,所述制备方法还包括后处理的步骤;
[0035]
所述后处理为:待s2步骤中的反应结束后,将反应物冷却至0℃-室温(25
±
5℃),而后进行离心提纯2-3次;
[0036]
进一步地,所述离心在反溶剂作用下进行;所述反溶剂选自乙酸甲酯、乙酸乙酯、叔丁醇中的一种或几种。在离心的过程中,反溶剂的使用很关键,特别是对于晶粒尺寸较小的情况,如果不使用反溶剂,很难实现纳米晶体的提纯;而当反溶剂如上所述时,效果最佳。
[0037]
作为本发明的较佳技术方案,所述制备方法包括如下步骤:
[0038]
(1)将碳酸铯、油酸和1-十八烯混合后在110-130℃下真空干燥0.8-1.2h,而后在惰性气氛下加热至140-160℃,待碳酸铯完全溶解,得到油酸铯前驱体溶液;
[0039]
将过渡金属卤化物、碘化铅、1-十八烯和三正辛基膦混合后在110-130℃下真空干燥0.8-1.2h,而后加入干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热至120-200℃,得卤化铅和过渡金属卤化物的混合溶液;
[0040]
(2)将所述油酸铯前驱体溶液与所述混合溶液快速混合,反应5s-5mins;待反应结束后,将反应物冷却至0℃-室温(25
±
5℃),而后进行离心提纯2-3次,即得过渡金属离子掺杂的钙钛矿量子点材料(即掺杂有过渡金属卤化物的钙钛矿量子点材料);
[0041]
其中,所述油酸铯前驱体溶液中,1-十八烯为溶剂,碳酸铯的浓度为10-30mg/ml,油酸的浓度为0.05-0.1ml/ml;所述混合溶液中,1-十八烯为溶剂,碘化铅的浓度为0.02-0.06mmol/ml,三正辛基膦的浓度为0.1-0.4ml/ml,油胺的浓度为0.1-0.4ml/ml,油酸的浓度为0.1-0.4ml/ml;以所述卤化铅的物质的量为基准,所述过渡金属卤化物的添加量为0.1-400%。
[0042]
进一步地,所述冷却采用水浴或冰水浴的方式。
[0043]
进一步地,所得的过渡金属离子掺杂的钙钛矿量子点材料分散保存在正己烷或甲
苯中。
[0044]
如此,本发明通过将特定的过渡金属离子掺杂到钙钛矿量子点材料中,可以有效改善因cs
半径较小引起的钙钛矿晶体结构相变的问题,提高全无机钙钛矿量子点材料的稳定性。
[0045]
基于上述方案,本发明的有益效果在于:
[0046]
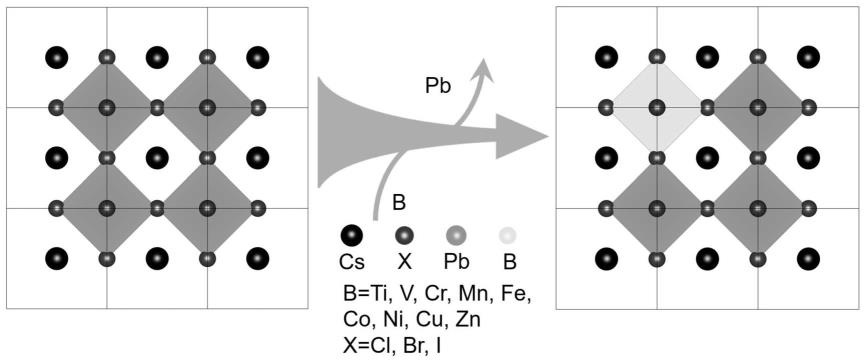
本发明通过在钙钛矿晶体结构中引入离子半径较小的过渡金属离子,显著提高了全无机钙钛矿量子点材料的晶体结构稳定性。以掺杂过渡金属镍为例,经过掺杂比例优化之后,掺杂过渡金属离子的钙钛矿量子点薄膜在85℃、85%相对湿度环境下存放100h之后仍然可以在365nm紫外灯照射下发出明亮的红光,其黑色相保留率超过95%,而没有掺杂的薄膜在40min保留了51.7%的黑色相,5h之后为17.3%,12h之后仅为0.34%,几乎完全失效,掺杂之后的稳定性提升了超过150倍;掺杂过渡金属离子的钙钛矿量子点薄膜直接浸泡在去离子水中,存放100h之后仍然具有优异的发光性能;掺杂过渡金属离子的钙钛矿量子点溶液与去离子水混合,搅拌200h之后仍然具有明亮的红色发光,284h之后还具有较高的发光强度。综上所述,掺杂之后的钙钛矿量子点湿热稳定性、水稳定性均显著提升。
[0047]
此外,本发明提供的钙钛矿量子点材料的制备方法,可通过改变反应温度、卤化铅与过渡金属卤化物的比例、反应时间、配体浓度等对晶粒尺寸和掺杂比例进行有效地调节,进而根据实际应用需要对反应产物性能进行调整;并且,该制备方法灵活,重复性高,反应参数调节简单高效。
附图说明
[0048]
图1为过渡金属离子掺杂的钙钛矿量子点材料的晶体结构示意图;
[0049]
图2为不同ni元素掺杂比例的cspbi3量子点xrd图谱;其中undoped代表不掺杂的cspbi3量子点,ni-5.28%代表实际产物中原子比ni/pb=5.28%,其它类似(注:ni/pb原子比例为电感耦合等离子体质谱仪测试结果);
[0050]
图3为cspbi3量子点透射电镜图;
[0051]
图4为不同ni掺杂比例的cspbi3量子点透射电镜图;其中,(a)、(b)、(c)、(d)分别对应ni-1.25%,ni-3.65%,ni-4.42%,ni-5.28%;
[0052]
图5为不同ni掺杂比例的cspbi3量子点吸收光谱和光致发光谱;
[0053]
图6为不同ni掺杂比例的cspbi3量子点薄膜在85℃、85%相对湿度条件下的时间分辨图;其中,左边为日光灯下的照片,右边为365nm紫外光激发下的照片;
[0054]
图7为根据图6日光灯下不同ni掺杂比例的滴涂薄膜黑色区域剩余比例绘制的云图;其中,100%表示黑色区域完全保留,薄膜样品没有发生相变;0%表示薄膜样品完全转变为非钙钛矿相;
[0055]
图8为掺杂样品(ni-3.65%)和非掺杂样品分别与去离子水混合之后的时间分辨图;其中,左为掺杂样品,右为非掺杂样品;测试过程通过搅拌将量子点溶液与去离子水混合;
[0056]
图9为掺杂样品(ni-3.65%)滴涂薄膜和非掺杂样品滴涂薄膜分别浸泡在去离子水中的时间分辨图;其中,左为掺杂样品,右为非掺杂样品。
具体实施方式
[0057]
以下实施例用于说明本发明,但不用来限制本发明的范围。
[0058]
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
[0059]
实施例1
[0060]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0061]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml 1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0062]
(2)制备碘化铅和碘化镍的混合溶液:将92.2mg碘化铅和62.5mg碘化镍称量并装入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到170℃;
[0063]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0064]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0065]
本实施例的钙钛矿量子点材料对应ni-1.25%样品。
[0066]
实施例2
[0067]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0068]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml 1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0069]
(2)制备碘化铅和碘化镍的混合溶液:将92.2mg碘化铅和187.5mg碘化镍称量并装入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到170℃;
[0070]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0071]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0072]
本实施例的钙钛矿量子点材料对应ni-3.65%样品。
[0073]
实施例3
[0074]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0075]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml 1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0076]
(2)制备碘化铅和碘化镍的混合溶液:将92.2mg碘化铅和187.5mg碘化镍称量并装
入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到185℃;
[0077]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0078]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0079]
本实施例的钙钛矿量子点材料对应ni-4.42%样品。
[0080]
实施例4
[0081]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0082]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml 1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0083]
(2)制备碘化铅和碘化镍的混合溶液:将92.2mg碘化铅和187.5mg碘化镍称量并装入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到200℃;
[0084]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0085]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0086]
本实施例的钙钛矿量子点材料对应ni-5.28%样品。
[0087]
实施例5
[0088]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0089]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0090]
(2)制备碘化铅和碘化钴的混合溶液:将92.2mg碘化铅和62.55mg碘化钴称量并装入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到170℃;
[0091]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0092]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0093]
实施例6
[0094]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0095]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全
溶解,将温度调回120℃,待后续使用;
[0096]
(2)制备碘化铅和碘化钴的混合溶液:将92.2mg碘化铅和125.10mg碘化钴称量并装入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到170℃;
[0097]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0098]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0099]
实施例7
[0100]
本实施例提供一种钙钛矿量子点材料,其制备方法包括如下步骤:
[0101]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml 1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0102]
(2)制备碘化铅和碘化钴的混合溶液:将92.2mg碘化铅和121.90mg碘化钒称量并装入25ml三口烧瓶,并加5ml 1-十八烯和1ml三正辛基膦,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到170℃;
[0103]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的混合溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0104]
(4)冷却后的反应物加入20ml乙酸甲酯,并采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0105]
图1为过渡金属离子掺杂的钙钛矿量子点材料的晶体结构示意图,其中,颜色最深的为cs,pb位于正八面体中心,过渡金属元素b部分替换正八面体中心的pb,形成稳定的晶体结构,卤素元素x分布在正八面体的6个顶角上;由此可以清晰地看出过渡金属离子的掺杂位点。
[0106]
对比例1
[0107]
本对比例提供一种钙钛矿量子点材料,该钙钛矿量子点材料为非掺杂的全无机钙钛矿cspbi3量子点,用以对比过渡金属离子掺杂后的稳定性提升效果;该钙钛矿量子点材料的制备方法包括如下步骤:
[0108]
(1)制备油酸铯前驱体溶液;将0.2g碳酸铯、0.7ml油酸和10ml 1-十八烯称量并装入25ml三口烧瓶,在120℃真空干燥1h之后,在惰性气体环境下加热到150℃,待碳酸铯完全溶解,将温度调回120℃,待后续使用;
[0109]
(2)制备碘化铅溶液:将92.2mg碘化铅称量并装入25ml三口烧瓶,并加5ml 1-十八烯,在120℃真空干燥1h之后,分别注入1ml干燥的油胺和油酸,待溶液澄清透明,在惰性气氛下加热到170℃;
[0110]
(3)取0.4ml步骤(1)所制备的油酸铯前驱体溶液快速注入步骤(2)的碘化铅溶液中,反应5秒,然后使用水浴将反应物冷却至室温;
[0111]
(4)冷却后的反应物采用8000rpm离心5分钟,倒掉上清液,使用1ml正己烷和2ml乙
酸甲酯重新分散沉淀;然后再使用8000rpm离心5分钟,倒掉上清液,使用2ml正己烷重新分散沉淀;最后1000rpm离心1分钟,保留上清液。
[0112]
本对比例的钙钛矿量子点材料对应undoped样品。
[0113]
试验例
[0114]
对实施例1-4、对比例1的钙钛矿量子点材料的性能进行测试,具体如下:
[0115]
湿热稳定性、水稳定性测试说明:
[0116]
85℃、85%相对湿度稳定性测试:85℃温度由加热板提供,85%相对湿度由湿度箱提供,通过加湿器维持湿度箱内稳定的湿度;测试时将滴涂的样品薄膜放置在加热板上,以时间为序,记录样品薄膜在日光灯和365nm紫外灯照射下的状态,进行湿热稳定性测试。
[0117]
量子点薄膜水稳定性测试:直接将滴涂的样品薄膜浸泡在去离子水中,以时间为序,记录样品薄膜在365nm紫外灯照射下的发光性能,完成薄膜水稳定性测试。
[0118]
量子点溶液水稳定性测试:将量子点溶液与等体积的去离子水混合,以时间为序,记录量子点溶液在365nm紫外灯照射下的发光性能,为保证量子点溶液与水充分混合,测试全程中使用磁力搅拌器对溶液进行搅拌。
[0119]
图2为不同ni元素掺杂比例的cspbi3量子点xrd图谱,其中undoped代表不掺杂的cspbi3量子点,ni-5.28%代表实际产物中原子比ni/pb=5.28%,其它类似(注:ni/pb原子比例为电感耦合等离子体质谱仪测试结果);由图2可知,实施例1-4、对比例1的钙钛矿量子点材料均为黑色相钙钛矿晶体结构。
[0120]
不同ni掺杂比例的cspbi3量子点的透射电镜图见图3和图4;由图3和图4可知,从undoped到ni-1.25%,再到ni-3.65%,随着前驱体溶液中nii2的增加,晶粒尺寸逐渐减小,掺杂比例逐渐提高;从ni-3.65%到ni-4.42%,再到ni-5.28%,随着反应温度的增加,晶粒尺寸逐渐增大,掺杂比例逐渐增大。
[0121]
图5为不同ni掺杂比例的cspbi3量子点吸收光谱和光致发光谱;由图5可知,吸收边和发光峰位置随掺杂比例的提高,先蓝移再红移,这与不同掺杂比例样品的晶粒尺寸相对应,cspbi3量子点半导体带隙会随着晶粒尺寸的变小而变大,进而导致发光峰位置蓝移。
[0122]
图6为不同ni掺杂比例的cspbi3量子点薄膜在85℃、85%相对湿度条件下的时间分辨图,其中,左图系列图片信息保存较为完整,左图中颜色越深代表黑色相保留程度越高,但是右图系列图片信息损失较多,右图中黑色区域应该为红色发光,并且ni-3.65%发出的红光最强,图片并没有很好展示,因此以左图了解主要信息;由图6可知,掺杂样品黑色相稳定性均大于非掺杂样品,ni-3.65%样品在经过100多个小时之后仍然能够发出鲜艳的红光。
[0123]
图7为根据图6日光灯下不同ni掺杂比例的滴涂薄膜黑色区域剩余比例绘制的云图;由图7可知,ni-3.65%和ni-4.42%两个样品在85℃、85%相对湿度条件下放置100h后,仍然具有高达95%的黑色相保留率,而不掺杂的样品在几个小时之内就已经基本失效,在第40min保留了51.7%,到第5h仅保留了17%黑色覆盖面积。
[0124]
图8为掺杂样品(ni-3.65%)与非掺杂样品与去离子水混合之后的时间分辨图;由图8可知,掺杂样品与去离子水混合之后仍然具有很强的稳定性,浸泡200h仍然具有明亮的发光。此外,图9为掺杂样品(ni-3.65%)滴涂薄膜和非掺杂样品滴涂薄膜分别浸泡在去离子水中的时间分辨图;由图9可知,掺杂样品滴涂薄膜的水稳定性也明显提高。需要特别说
明的是,图8和图9为样品在365nm紫外光照射下的图片,亮度越高代表样品发光性能越好。
[0125]
并且,实施例5-7的钙钛矿量子点材料能够实现与上述实施例1-4相同的技术效果。
[0126]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。