制备方法及其作为声敏剂的应用。该铜卟啉-叶酸脂质体纳米颗粒用叶酸靶向的脂质体为载 体,提高了水溶性和靶向性,有利于在肿瘤部位富集,进而提高sdt治疗效果。但是该 研究对于卟啉类化合物的光毒性没有进行研究。
5.声敏剂的原理是在超声的靶向作用下产生氧活性物质(包括单线态氧),对靶向的癌细 胞产生杀伤作用。目前,常用的声敏剂是卟啉及卟啉衍生物,但是这类声敏剂本身从光敏 剂发展而来,具有一定的光敏性,使得患者在接受声敏剂治疗的同时需要严格避光,且接 受治疗后的一段时间也要避光处理,造成治疗的不便利。与此同时,该类声敏剂的光毒性 易导致严重的皮肤毒副作用。因此,研发一种具有低的光毒性声敏剂对于声敏剂的发展和 临床应用具有重要的意义。
技术实现要素:
6.相比现有技术,本发明提供了一种分子结构新颖,低光毒性,生物安全性高,sdt功 效强的新型有机小分子声敏剂。这种新型小分子作为光敏剂在肿瘤声动力治疗方面具有广 阔的应用前景。此外还可以和其他诊疗手段进行联合,比如化疗,超声靶向微泡破坏技术 (utmd),磁共振成像(mri),近红外荧光成像(nir-fli)等,以期得到更好的治 疗效果。
7.为实现上述目的,本发明提供了以下技术方案:
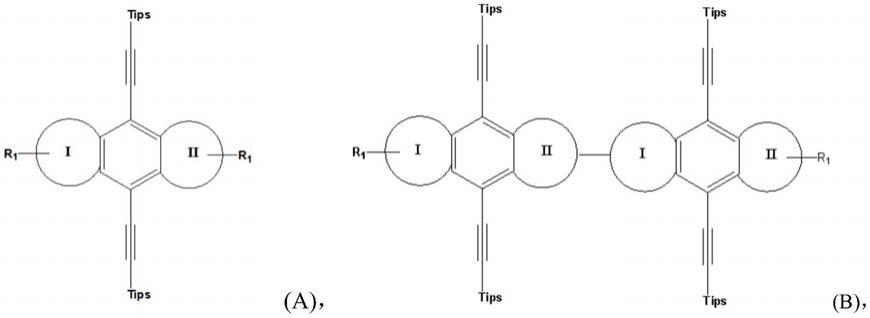
8.本发明第一个目的是提供一种具有低光毒性的声敏剂,具有如下式a或式b的结构 简式:
[0009][0010]
其中环i,环ii独立地选自芳香族稠合环,具体选自苯基,萘基,蒽基,联苯基,呋 喃基,噻吩基,硒吩基。;tips为硅烷基;r1独立地为,h,c1-6烷基、c1-6烯基,c1-6 炔基,羟基、硝基,其中*表示和芳香族稠合环i或ii相邻两个碳原子链接的键,条件是r 1
中至少一
个为r2独立地为h,c1-6烷基,c1-6烯基,c1-6炔基,羟基、 硝基,n为1-5之间的整数,比如1,2,3,4,5。
[0011]
所述芳香族稠合环选自苯基,萘基,蒽基,联苯基,呋喃基,噻吩基、苯并呋喃基、 苯并噻吩基;所述硅烷基选自-si[ch(ch3)2]3、-si[ch2ch3]3或-si[c(ch3)3]3。
[0012]
发明人预料不到地发现,具有含有噻吩基的上述式a结构的化合物具有较强的声敏 性,同时具有较低的光毒性,适合作为声动力治疗的声敏剂使用。此外,具有式a结构或 者类似结构的二聚体,比如式b和式c化合物,具有较低的光毒性。所述低毒性是指在可 见光照射下所产生的ros不足以造成细胞死亡(ic
50
>200μg/ml)。此类化合物由于合成的 便利性,结构可控和方便引入各种基团,有望开发一类新的具有临床应用价值的声敏剂。
[0013]
进一步地,本发明所述小分子光敏剂为以下化合物1-5:
[0014][0015]
本发明第二个目的是提供所述小分子光敏剂的制备方法,包括以下步骤:
[0016]
(1)噻吩-2,3-二甲醛和1,4-二羟基芳香族化合物在碱性条件下反应得到含有噻唑基和 苯醌基的中间产物i;
[0017]
(2)三异丙基硅基乙炔加入溶剂,在低温条件下混合,逐滴加入烷基锂和中间产物i, 升温后反应15-30h,加入二价锡盐,萃取、干燥,提纯得到噻吩并芳香化合物前体;
[0018]
(3)噻吩并芳香化合物前体在二异丙基氨基锂存在下和二(三甲苯基)氟化硼反应经过后 处理得到式a化合物;或者溴代噻吩并芳香化合物前体在双联频哪醇硼酸酯,以及碳酸钠 和钯催化剂存在下反应得到二聚体,进而二聚体在二异丙基氨基锂存在下和二(三甲苯基) 氟化硼反应经过后处理得到式b化合物。
[0019]
其中溴代的噻吩并芳香化合物前体是步骤(1)中采用溴代的1,4-二羟基芳香族化合物为 原料按照相同的操作和条件得到。
[0020]
进一步地,步骤(1)中碱性条件是加入氢氧化钠和/或氢氧化钾,噻吩-2,3-二甲醛和1,4
‑ꢀ
二羟基芳香族化合物的摩尔比为1:1-1.5。
[0021]
进一步地,步骤(2)中,所述溶剂为四氢呋喃,乙醚,正己烷,甲苯中的至少一种;所 述低温是冷却至零下50℃至零下80℃,所述烷基锂选自正丁基锂,所述二价锡盐为二水 合氯化锡(ii);中间产物i,三异丙基硅基乙炔,烷基锂的摩尔比为1:2-4:2-4。
[0022]
进一步地,步骤(3)中,当制备式a化合物时,噻吩并芳香化合物前体,二异丙基氨 基锂和二(三甲苯基)氟化硼的摩尔比为1:2-3:2-3;制备式b化合物时,溴代噻吩并芳香 化合物前体、双联频哪醇硼酸酯、碳酸钠、钯催化剂的摩尔比为1:0.5-0.8:5-8:0.05-0.1。
[0023]
本发明第三个目的是提供一种上述述光敏剂在制备声动力治疗药物中用途。具体操作 是0.3-3w/cm2条件对患者进行治疗,声敏剂剂量为0.5mg/kg
[0024]
相对于现有技术,本发明取得了以下有益效果:
[0025]
一、本发明提供了一种结构新颖的小分子声敏剂,具有较低的光毒性(ic
50
>200μg/ml), 同时具有较强的声毒性(ic
50
<2μg/ml),在进行声动力治疗时,避免了一般声敏剂具有较强 的光毒性带来的需要避光等弊端,是一种具有临床应用潜力的声敏剂药物。
[0026]
二、本发明声敏剂结构易于修饰,合成方法简单,可以通过丰富的合成策略得到不同 结构的声敏剂。
附图说明
[0027]
图1是化合物1的质谱图;
[0028]
图2是化合物2的质谱图;
[0029]
图3是化合物1和2在530nm处在光照(pdt)和超声(sdt)荧光强度;
[0030]
图4是化合物1和2在不同浓度下的细胞存活率;
[0031]
图5是本发明化合物和二氢卟吩的体外声毒性测试结果;
[0032]
图6是接种肿瘤细胞小鼠的体重和时间的关系变化图;
[0033]
图7是接种肿瘤细胞小鼠的肿瘤体积和时间的关系变化图。
具体实施方式
[0034]
以下结合实施例对本发明进行进一步的说明。
[0035]
制备例1化合物1的制备
[0036][0037]
1)在氮气保护下,将噻吩-3-甲醛(1.0当量)、乙二醇(5.0当量)和对甲苯磺酸(1% 当量)的原料溶解在无水甲苯中,在130℃下反应9小时,得到2-(3-噻吩基)-1,3-二 氧戊环。
[0038]
2)向烘箱干燥的schlenk烧瓶中加入2-(3-噻吩基)-1,3-二氧戊环(1.0当量)。然 后加入无水无氧的四氢呋喃并将混合物冷却至-78℃。通过注射器逐滴加入正丁基锂 (1.1当量)。将该溶液在-78℃下搅拌15分钟,然后加入n,n-二甲基甲酰胺(2.3 当量),使溶液升温至室温并继续搅拌过夜。然后加入3m盐酸水溶液,将所得混合 物在加热回流搅拌下30分钟后在60℃搅拌过夜并用二氯甲烷萃取。有机层用盐水洗 涤并经无水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,以石油醚和 乙酸乙酯为洗脱剂,得到噻吩-2,3-二甲醛。
[0039]
3)将噻吩-2,3-二甲醛(1.0当量)和1,4-二羟基萘(1.0当量)溶解在四氢呋 喃和乙醇(2:1)的热混合溶液中。然后在搅拌的同时加入几滴15%氢氧化钠水溶液 直到观察到沉淀。将该溶液搅拌一小时,随后过滤,用甲醇洗涤直至滤液无色,然后 用四氢呋喃和乙醚洗涤得到蒽[2,3-b]噻吩-5,10-二酮。
[0040]
4)向烘箱干燥的schlenk烧瓶中加入三异丙基硅基乙炔(3.5当量)。然后加入无水 无氧四氢呋喃并将混合物冷却至-78℃。通过注射器逐滴加入正丁基锂(3.4当量)。 将该溶液在-78℃下搅拌1小时,然后加入蒽[2,3-b]噻吩-5,10-二酮(1.0当量),使溶 液升温至室温并继续搅拌过夜。然后加入二水合氯化锡(ii)在10%盐酸水溶液中的 饱和溶液,将所得混合物在室温下搅拌1小时并用二氯甲烷萃取。有机层用盐水洗涤 并经无水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,以石油醚为洗 脱剂,得到噻吩并蒽前体。
[0041]
5)向schlenk烧瓶中加入噻吩并蒽前体(1.0当量),然后加入四氢呋喃并将混合 物冷却至-78℃。通过注射器逐滴加入二异丙基氨基锂(2.1当量)。将该溶液在-78℃ 下搅拌5小时,然后加入二(三甲苯基)氟化硼(2.2当量),使溶液升温至室温并继续 搅拌过夜。通过加入水淬灭反应。混合物用二氯甲烷萃取,有机层用盐水洗涤并经无 水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,得到硼杂噻吩并蒽终 产物1。
[0042]
制备例2化合物2的制备
[0043][0044]
1)将噻吩-2,3-二甲醛(1.0当量)和1,4-二羟基蒽(1.0当量)溶解在四氢呋 喃和乙醇(2:1)的热混合溶液中。然后在搅拌的同时加入几滴15%氢氧化钠水溶液 直到观察到沉淀。将该溶液搅拌一小时,随后过滤,用甲醇洗涤直至滤液无色,然后 用四氢呋喃和乙醚洗涤得到并四苯并[2,3-b]噻吩-5,12-二酮。
[0045]
2)向烘箱干燥的schlenk烧瓶中加入三异丙基硅基乙炔(3.5当量)。然后加入无水 无氧四氢呋喃并将混合物冷却至-78℃。通过注射器逐滴加入正丁基锂(3.4当量)。 将该溶液在-78℃下搅拌1小时,然后加入并四苯并[2,3-b]噻吩-5,12-二酮(1.0当量), 使溶液升温至室温并继续搅拌过夜。然后加入二水合氯化锡(ii)在10%盐酸水溶液 中的饱和溶液,将所得混合物在室温下搅拌1小时并用二氯甲烷萃取。有机层用盐水 洗涤并经无水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,以石油醚 为洗脱剂,得到噻吩并四苯前体。
[0046]
3)向schlenk烧瓶中加入噻吩并四苯前体(1.0当量),然后加入四氢呋喃并将混 合物冷却至-78℃。通过注射器逐滴加入二异丙基氨基锂(2.1当量)。将该溶液在
ꢀ‑
78℃下搅拌5小时,然后加入二(三甲苯基)氟化硼(2.2当量),使溶液升温至室温并 继续搅拌过夜。通过加入水淬灭反应。混合物用二氯甲烷萃取,有机层用盐水洗涤并 经无水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,得到硼杂噻吩并 四苯终产物2。
[0047]
对目标化合物1和2进行高分辨率maldi-tof质谱分析,其结果如图1和2所示, 化合物1m/z=842.4727([m] ,100%,c
56h71
b1s1si2的计算值为842.4908);化合物2m/z =892.4908([m] ,100%,c
60h73
b1s1si2的计算值为892.5065)。其分子量与同位素的峰 形均与理论值一致。
[0048]
制备例3化合物5的制备
[0049][0050]
基本操作和制备例2相同,区别在于在得到首先制备溴代噻吩并蒽前体后,加入双联 频哪醇硼酸酯,醋酸钯和碳酸钠反应得到二聚体。进而二聚体在二异丙基氨基锂存在下和 二(三甲苯基)氟化硼反应得到化合物5。
[0051]
具体操作是:
[0052]
1)向schlenk烧瓶中加入4-溴-1,2-二甲苯(1.0当量),n-溴代琥珀酰亚胺(4.0当量) 和过氧化苯甲酰(0.1当量),然后加入无水无氧四氯化碳并将混合物升温至80℃,搅拌过 夜。接下来降温至0℃,将不溶物滤出,得到粗产物4-溴-1,2-双(二溴甲基)苯。
[0053]
2)向schlenk烧瓶中加入4-溴-1,2-双(二溴甲基)苯(1.0当量),苯醌(7.0当量)和 碘化钾(4.0当量),然后加入无水无氧n,n-二甲基乙酰胺并将混合物升温至110℃,搅拌过 夜。接下来倒入水和甲醇混合溶液中,搅拌过滤。过滤物用二氯甲烷萃取,有机层用盐水 洗涤并经无水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,用石油醚和二 氯甲烷作为洗脱剂,得到6-溴蒽-1,4-二酮。
[0054]
3)向schlenk烧瓶中加入6-溴蒽-1,4-二酮(1.0当量)和连二亚硫酸钠(4.0当量),然 后加入二恶烷与水的混合溶液并将混合物室温搅拌4小时。接下来再次加入连二亚硫酸钠 (2.0当量),室温搅拌4小时。接下来倒入水中,搅拌过滤。过滤物用水洗涤干燥得到6
‑ꢀ
溴蒽-1,4-二醇。
[0055]
4)将噻吩-2,3-二甲醛(1.0当量)和6-溴蒽-1,4-二醇(1.0当量)溶解在四氢呋喃 和乙醇(2:1)的热混合溶液中。然后在搅拌的同时加入几滴15%氢氧化钠水溶液直到观察 到沉淀。将该溶液搅拌一小时,随后过滤,用甲醇洗涤直至滤液无色,然后用四氢呋喃和 乙醚洗涤得到8-溴并四苯[2,3-b]噻吩-5,12-二酮。
[0056]
5)向烘箱干燥的schlenk烧瓶中加入三异丙基硅基乙炔(3.5当量)。然后加入无水无 氧四氢呋喃并将混合物冷却至-78℃。通过注射器逐滴加入正丁基锂(3.4当量)。将该溶 液在-78℃下搅拌1小时,然后加入8-溴并四苯并[2,3-b]噻吩-5,12-二酮(1.0当量),使溶 液升温至室温并继续搅拌过夜。然后加入二水合氯化锡(ii)在10%盐酸水溶液中的饱和 溶液,将所得混合物在室温下搅拌1小时并用二氯甲烷萃取。有机层用盐水洗涤并经无水 硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,以石油醚为洗脱剂,得到溴 代噻吩并四苯前体。
[0057]
6)向schlenk烧瓶中加入溴代噻吩并蒽前体(1.0当量),双联频哪醇硼酸酯(0.5当 量),醋酸钯(0.1当量)和碳酸钠(8.0当量),然后加入无水无氧二氧杂环己烷并将混合物升 温至80℃,搅拌过夜。通过加入水淬灭反应。混合物用二氯甲烷萃取,有机层用盐水洗涤 并经无水硫酸钠干燥。旋转蒸发后,粗产物通过柱色谱法(硅胶)纯化,用石油醚和二氯甲 烷作为洗脱剂,得到二聚体。
[0058]
7)向schlenk烧瓶中加入二聚体(1.0当量),然后加入四氢呋喃并将混合物冷却至
ꢀ‑
78℃。通过注射器逐滴加入二异丙基氨基锂(3.0当量)。将该溶液在-78℃下搅拌5小 时,然后加入二(三甲苯基)氟化硼(3.2当量),使溶液升温至室温并继续搅拌过夜。通过 加入水淬灭反应。混合物用二氯甲烷萃取,有机层用盐水洗涤并经无水硫酸钠干燥。旋转 蒸发后,粗产物通过柱色谱法(硅胶)纯化,得到硼杂噻吩并四苯二聚体终产物5。
[0059]
化合物4根据化合物5类似的方法制得,区别在于溴代的噻吩并蒽前体不同。
[0060]
实施例1
[0061]
1)将化合物1和2溶于有机溶剂中,加入亲水性物质peg和水,混合均匀后得到混 合溶液,减压旋蒸出有机溶剂,得到亲水性1和2储备液。
[0062]
2)活性氧(ros)可由绿色荧光探针2
′
,7
′–
二氯二氢荧光素二乙酸酯(dcfh-da)特异 性检测。将10mm的1或2储备液加入到比色皿中,并加入dcfh-da探针,分别进行超 声或光照15分钟,在此期间每3分钟对530nm波长处荧光强度进行检测,作为产生活性 氧的标志。
[0063]
结果如图3所示,在530nm处绘制出荧光强度增加随作用时间的变化趋势。光照(pdt) 后,dcfh-da的荧光微弱且强度保持不变,而超声(sdt)的荧光强度随超声时间的延长逐 渐增加,化合物1和2在超声作用下生成ros是导致荧光强度增加的原因。说明本发明 化合物1和2有较强的声敏性,而具有较低的光毒性。
[0064]
实施例2
[0065]
将制备得到的化合物1、2溶解于pbs液中,并用pbs溶液将浓度稀释,得到0、0.2、 0.5、1、2、5、10、20ug/ml的声敏剂制剂。将不同浓度制剂加入细胞,培养24小时后, 检测细胞活性。结果如图4所示,在没有超声的情况下,随着1和2浓度的增加,观察 到的细胞存活率与空白对照组保持一致,表明其在细胞中具有良好的生物相容性,无细胞 毒性。
[0066]
实施例3
[0067]
将制备得到的化合物1、2溶解于pbs液中,并用pbs溶液将浓度稀释,得到0、0.2、 0.4、0.6、0.8、1.0、2.0ug/ml的声敏剂制剂。将不同浓度制剂及2.0ug/ml的声敏剂制剂 ce6分别加入细胞,培养6小时后,将细胞用超声(1w cm2,3分钟)进行刺激,再孵育24小时,检测细胞活性,评估体外sdt效果,结果如图5所示,其中us代表只有超声 作为空白组,ce6为二氢卟吩,在超声刺激下,空白组(us)的细胞存活率没有显著下降, 而化合物1和化合物2治疗组的细胞存活率显著降低。特别是2ug/ml浓度的细胞存活率 小于20%,而相同浓度的ce6治疗组的细胞存活率接近90%,表明1和2对肿瘤细胞的 sdt治疗是非常高效的。
[0068]
该专利所述的其余化合物均进行了相同的实验,所得到的光动力和声动力治疗效果如 表1所示,结果表明该专利所涉及的化合物均具有较低的光毒性和较好的声动力治疗效果。
[0069]
实施例4
[0070]
对本发明实施例制得化合物1-5以及二氢卟吩ce6进行光毒性(白炽灯,100w,3分 钟)和声毒性(超声仪,1w cm2,3分钟)的测试,测试方法是根据细胞实验结果,以存 活率为纵坐标,浓度为横坐标,使用spss21.0对结果进行回归计算,取结果中50%规整 率的数值为ic
50
,结果如下表1所示。
[0071]
表1化合物光毒性和声毒性测试。
[0072]
化合物光动力活性ic
50
/μg/ml声动力活性ic
50
/μg/ml化合物1>2000.96化合物2>2000.72化合物3>2000.84化合物4>2001.03化合物5>2000.96ce676.984.7
[0073]
说明本发明制得化合物具有低的光毒性和高的声毒性,是一种极具潜力的临床声敏 剂。
[0074]
实施例5
[0075]
在小鼠背部皮下注射100μl含1
×
105个4t1肿瘤细胞的磷酸盐缓冲液。当肿瘤体积接 近70mm3时,对小鼠进行治疗。将小鼠随机分为4组(每组5只):pbs组(对照组,只注 射了磷酸盐缓冲溶液);2组(化合物2的浓度0.2mg/ml注射入患有背部瘤的裸鼠体内);2 us组(化合物2的浓度0.2mg/ml注射入患有背部瘤的裸鼠体内,同时伴随超声);us 组(只有超声)。将浓度为0.2mg/ml的2制剂注射入患有背部瘤的裸鼠体内,24小时后 将超声组的小鼠进行肿瘤超声(1w cm2,3分钟)治疗,每隔两天测量小鼠重量和肿瘤体 积,14天后评估治疗效果。治疗后每隔两天监测各组肿瘤体积和小鼠体重,各组均可观察 到小鼠体重的增长(图6),表明化合物2在体内具有良好的生物相容性和低毒性。
[0076]
图7是pbs组,2组,2 us组,us组的小鼠每隔两天监测各组肿瘤体积,其中pbs 组、us组,2组治疗均表现出相似的肿瘤生长速率,但2 us组前6天内得到明显抑制, 小鼠肿瘤未复发,表明化合物2能够有效抑制了肿瘤,证明了化合物2的sdt治疗具有 显著效果。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。