1.本发明涉及一种热激活延迟荧光材料及其应用,属于有机发光材料领域,特别涉及一类含有酚氧-吡啶螯合二氟化硼受体结构单元的热激活延迟荧光有机发光材料,以及包含该热激活延迟荧光有机发光材料的有机电致发光器件。
背景技术:
2.有机发光二极管(organic light-emitting diode,oled)近些年取得了突飞猛进地发展,相比于lcd显示技术,oled具有如下优势:自主发光,无需背光源,节能;可折叠或弯曲;响应速度快,对比度高;工作温度范围宽;材料成本低、发光效率高等。oled已成为新一代的全彩显示和照明技术,受到学术界和工业界的广泛重视,其在平板显示、固体照明、军事和航空航天领域得到了广泛的应用,并且有望在未来取代液晶显示器(liquid crystal display,lcd)。在oled发展初期,主要采用传统有机小分子荧光材料,该材料具有很好的器件稳定性。但是其理论上最多只能利用25%的单重态激子,剩余75%的三重态激子因跃迁禁阻只能通过非辐射跃迁失活,限制了其在oled中的应用。第二代磷光材料由于其重金属的自旋轨道耦合效应(soc),可以打破三线态跃迁禁阻,能有效促进电子由单线态到三线态的系间蹿越,充分利用电致激发产生的所有单线态和三线态激子,使其最大理论量子效率达100%。磷光材料通常需要引入极为昂贵的稀有重金属,其制备成本较高,且贵金属储量有限,制约了第二代oled器件的发展。而后日本九州大学adachi教授合成了一系列给-受体骨架的热激活延迟荧光(tadf,thermally activated delayed fluorescence)材料,这种给体-受体分子设计能实现homo和lumo的空间分离,达到较小的homo和lumo重叠,具有较小的单线态和三线态能级差δe
s1-t1
。在周围环境中热量的激发下能够实现从最低三重激发态(t1)态到最低单重激发态(s1)的反向系间窜越(risc),从而有效利用三线态激子产生延迟荧光,其理论内量子效率也可达到100%。tadf材料最大的优点就是采用纯有机材料,避免了使用含贵重金属的金属有机配合物,在oled领域有巨大的应用前景,并且基于tadf的oled取得了巨大进步。然而,长寿命和高亮度的高效tadf材料的设计仍然具有挑战性,有机硼化合物具有独特的光学和电子特性,被广泛研究并用作有机光电子材料,有望实现高效稳定发光,解决上述问题。同时,新型高效的蓝光热致延迟荧光材料的设计和开发亦是oled领域的重大问题。
技术实现要素:
3.本发明的目的在于提供一种基于酚氧-吡啶螯合二氟化硼受体结构单元构建的给-受体型热激活延迟荧光材料,所述的材料可用于oled显示和照明领域。
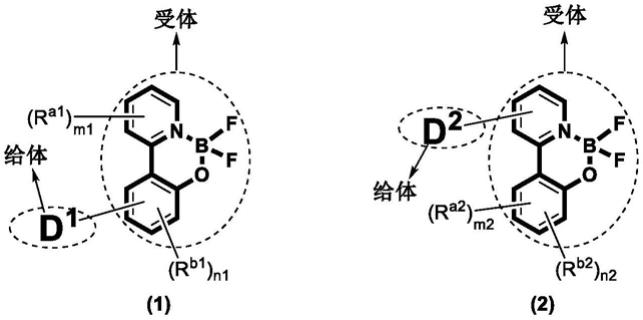
4.其特征在于,其化学式如通式(1)或(2)所示:
[0005][0006]
其中,
[0007]
式(1)中,r
a1
、r
b1
各自独立地为氢、氘、c
1-c
24
的烷基、c
1-c
24
的烷氧基、c
3-c
24
的环烷基、c
1-c
24
的醚、c
1-c
24
的杂环基、c
4-c
24
的芳基、c
4-c
24
的芳氧基、卤素、单或二烷基氨基、单或二芳基氨基、氰基或其组合;
[0008]
m1、n1分别为r
a1
和r
b1
的个数;其中,m1为0-4的整数,n1为0-3的整数;
[0009]
式(2)中,r
a2
、r
b2
各自独立地为氢、氘、c
1-c
24
的烷基、c
1-c
24
的烷氧基、c
3-c
24
的环烷基、c
1-c
24
的醚、c
1-c
24
的杂环基、c
4-c
24
的芳基、c
4-c
24
的芳氧基、卤素、单或二烷基氨基、单或二芳基氨基、氰基或其组合;
[0010]
m2、n2分别为r
a2
和r
b2
的个数;m2为0-4的整数,n2为0-3的整数;
[0011]
给体d1、d2各自独立地为以下结构之一:
[0012][0013]
其中,
[0014]
r1、r2、r3、r4、r7、r8、r
10
、r
11
、r
12
、r
13
、r
14
和r
15
各自独立地为氢、氘、c
1-c
24
的烷基、c
1-c
24
的烷氧基、c
3-c
24
的环烷基、c
1-c
24
的醚、c
1-c
24
的杂环基、c
4-c
24
的芳基、c
4-c
24
的芳氧基、卤素、硅基、单或二烷基氨基、单或二芳基氨基、氰基或其组合,其中相邻的两个取代基可以稠合成环;
[0015]
o1、p1、q1、r1、s1、t1、u1、v1、w1、x1、y1和z1分别为r1、r2、r3、r4、r7、r8、r
10
、r
11
、r
12
、r
13
、r
14
和r
15
的个数;
[0016]
o1和p1是0-5的整数;q1、r1、s1、t1、u1、v1、w1、x1、y1和z1是0-4的整数。
[0017]
进一步,所述热激活延迟荧光材料的结构式可优选为如(3)和(4)所示:
[0018][0019]
其中,
[0020]
式(3)中,r
a3
、r
b3
各自独立地为氢、氘、c
1-c
24
的烷基、c
1-c
24
的烷氧基、c
3-c
24
的环烷基、c
1-c
24
的醚、c
1-c
24
的杂环基、c
4-c
24
的芳基、c
4-c
24
的芳氧基、卤素、单或二烷基氨基、单或二芳基氨基、氰基或其组合;
[0021]
m3、n3分别为r
a3
和r
b3
的个数;其中,m3为0-4的整数,n3为0-3的整数;
[0022]
式(4)中,r
a4
、r
b4
各自独立地为氢、氘、c
1-c
24
的烷基、c
1-c
24
的烷氧基、c
3-c
24
的环烷基、c
1-c
24
的醚、c
1-c
24
的杂环基、c
4-c
24
的芳基、c
4-c
24
的芳氧基、卤素、单或二烷基氨基、单或二芳基氨基、氰基或其组合;
[0023]
m4、n4分别为r
a4
和r
b4
的个数;m4为0-4的整数,n4为0-3的整数;
[0024]
所述的给体d3为下列结构之一:
[0025][0026]
其中,
[0027]r1'
、r
2'
、r
3'
、r
4'
、r
7'
、r
8'
、r
10'
、r
11'
、r
12'
、r
13'
、r
14'
和r
15'
各自独立为氢、氘、c
1-c
24
的烷基、c
1-c
24
的烷氧基、c
3-c
24
的环烷基、c
1-c
24
的醚、c
1-c
24
的杂环基、c
4-c
24
的芳基、c
4-c
24
的芳氧基、卤素、硅基、单或二烷基氨基、单或二芳基氨基、氰基或其组合,其中相邻的两个取代基可以稠合成环;
[0028]
o2、p2、q2、r2、s2、t2、u2、v2、w2、x2、y2和z2分别为r1’
、r2’
、r3’
、r4’
、r7’
、r8’
、r
10’、r
11’、r
12’、r
13’、r
14’和r
15’的个数;
[0029]
o2和p2是0-5的整数;q2、r2、s2、t2、u2、v2、w2、x2、y2和z2是0-4的整数。
[0030]
进一步,本发明所述的热激活延迟荧光材料优选为如下之一的结构:
[0031]
[0032]
[0033][0034]
本发明所使用的术语“杂芳基”包括氮杂环丁烷基、二噁烷基、呋喃基、咪唑基、异噻唑基、异噁唑基、吗啉基、噁唑基(1,2,3-噁二唑基、1,2,5-噁二唑基和1,3,4-噁二唑基)
哌嗪基、哌啶基、吡嗪基、吡唑基、哒嗪基、吡啶基、嘧啶基、吡咯基、吡咯烷基、四氢呋喃基、四氢吡喃基、1,2,4,5-四嗪基、1,2,3,4-四唑基、1,2,4,5-四唑基、1,2,3-噻二唑基、1,2,5-噻二唑基、1,3,4-噻二唑基、噻唑基、噻吩基、1,3,5-三嗪基、1,2,4-三嗪基、1,2,3-三唑基、1,3,4-三唑基等。
[0035]
本发明使用的术语“甲硅烷基”通过式—sir1r2r3表示,其中r1,r2和r3可独立地为氢或者本发明所述的烷基、环烷基、烷氧基、烯基、环烯基、炔基、环炔基、芳基,或者杂芳基。
[0036]
本发明使用的“r
a1
,”“r
a2
,”“r
a3
,”“r
an”(其中n为整数)可独立地具有所列举的基团中的一个或者多个。例如,如果r1为直链烷基,那么烷基的一个氢原子可任选取代有羟基、烷氧基、烷基、卤素等。取决于选择的基团,第一基团可结合在第二基团内,或者可选择地,第一基团可悬挂(即,连接)至第二基团。
[0037]
本发明所述化合物可含有“任选取代的”部分。通常,术语“取代的”(无论在前面是否存在术语“任选”)意味着指出的部分的一个或者多个氢被适合的取代基替代。除非另作说明,否则“任选取代的”基团可在基团的每个可取代位置具有适合的取代基,以及当在任何给出的结构中超过一个位置可取代有超过一个选自指定基团的取代基时,在每个位置的取代基可相同或者不同。本发明设想的取代基组合优选为形成稳定的或者化学上可行的化合物的那些。在某些方面,除非清楚地相反指示,否则还涵盖的是,各个取代基可进一步任选被取代(即,进一步取代或未取代的)。
[0038]
化合物的结构可通过下式表示:
[0039][0040]
其被理解为等同于下式:
[0041][0042]
其中m通常为整数。即,ra被理解为表示五个单独的取代基r
a(1)
,r
a(2)
,r
a(3)
,r
a(4)
,r
a(5)
。“单独的取代基”是指每个ra取代基可独立地限定。例如,如果在一个情况中r
a(1)
为卤素,那么在这种情况下r
a(2)
不一定是卤素。
[0043]
在本发明中,基于含有酚氧-吡啶螯合二氟化硼受体结构单元的热激活延迟荧光材料为电中性。
[0044]
本发明的提供的含有酚氧-吡啶螯合二氟化硼受体结构单元的热激活延迟荧光材料可以有多种用途,既可用作有机电致发光器件的发光材料,亦可作为主体材料或其他功能层材料,可应用于全彩显示器、照明器件等。
[0045]
在本发明涉及的光学或电光装置,其包含上述含有酚氧-吡啶螯合二氟化硼受体结构单元的热激活延迟荧光材料的一种或多种。
[0046]
与现有技术相比,本发明的有益效果在于:
[0047]
酚氧-吡啶螯合二氟化硼受体结构单元的tadf材料,硼原子先与苯酚基相连,空的p轨道再与吡啶n原子上的孤对电子对配位,形成稳定的四配位结构。四配位硼化合物具有
良好的化学和热稳定性,尤其是含有b-o和b-f键的化合物由于其高的键解离能(分别为536和613kj/mol)。苯酚基螯合二氟化硼与吡啶配位,电子离域到整个共轭体系,使吡啶的π电子接受能力得到提升,再通过引入具有π共轭体系的给电子基团来构建具有给体-受体结构的分子,促进分子内电荷转移(ict,intramolecular charge transfer),获得tadf效应。此外还能形成并环结构的刚性π共轭骨架,能够有效抑制振动耦合导致的能量损失,提高发光量子效率。在电子受体和给体之间的引入取代基,可以有效调节给受体之间的二面角,打破共轭实现光谱蓝移。并且还能实现homo与lumo的不同程度分离,可以获得较小的δe
s1-t1
,实现tadf发光。酚氧-吡啶螯合二氟化硼受体结构单元的tadf材料具有很强的发光效率和较高的载流子迁移率,其光电性能强烈依赖于配体性质,可以通过配体的设计有效调控其光物理性质,是一种很有前途的发光材料。
附图说明
[0048]
图1分别是发光材料p-cz-bf2、p-tbucz-bf2、p-phcz-bf2、p-dmac-bf2、p-dpa-bf2、p-ptz-bf2、p-pxz-bf2、m-cz-bf2和m-phcz-bf2的甲苯溶液在室温下的发射光谱谱图。
[0049]
图2是发光材料p-cz-bf2、p-tbucz-bf2、p-phcz-bf2、p-dmac-bf2、p-dpa-bf2、p-ptz-bf2、p-pxz-bf2、m-cz-bf2和m-phcz-bf2的depeo薄膜在室温下的发射光谱谱图。
[0050]
图3是发光材料p-cz-bf2、p-tbucz-bf2、p-phcz-bf2、p-dmac-bf2、p-dpa-bf2、p-ptz-bf2、p-pxz-bf2、m-cz-bf2和m-phcz-bf2的depeo薄膜发光衰减(归一化发光强度-时间)曲线。
[0051]
图4是发光材料p-cz-bf2作为发光体在双主体材料不同比例及电子传输材料不同掺杂的器件电致发光谱图。
[0052]
图5是发光材料p-cz-bf2作为发光体在双主体材料不同比例及电子传输材料不同掺杂的器件电流密度-电压-发光强度曲线。
[0053]
图6是发光材料p-cz-bf2作为发光体在双主体材料不同比例及电子传输材料不同掺杂的器件外部量子效率-电流密度曲线。
[0054]
图7是发光材料m-phcz-bf2作为发光体在不同功能层厚度下的器件电致发光谱图。
[0055]
图8是发光材料m-phcz-bf2作为发光体在不同功能层厚度下的器件电流密度-电压-发光强度曲线。
[0056]
图9是发光材料m-phcz-bf2作为发光体在不同功能层厚度下的器件外部量子效率-电流密度曲线。
具体实施方式
[0057]
以下实施例向本领域普通技术人员提供如何制造和评价本发明描述的化合物及其oled器件,所述实施例仅是本公开内容的示范且不圈定限制范围。尽管已经尽力确保关于数值(例如,量、温度等)的准确性,但是应当考虑一些误差和偏差。除非另外说明,否则温度是以℃为单位或者在环境温度下,且压力是在大气压下或附近。
[0058]
本实施例中所叙述的用于本发明所描述的公开化合物的制备方法是众多方法中的一种,本发明申请公开化合物的制备方法尚有很多其他方法,本技术不圈定限制范围。因
此,本公开内容所属领域的技术人员可容易地修改所叙述的方法或者利用不同的方法来制备所公开的化合物的一种或多种。下列方法仅是示例性的,温度、催化剂、浓度、反应物组成、以及其它工艺条件可改变,并且对于期望的化合物,本公开内容所属领域的技术人员可以容易的选择合适的反应物和条件进行制备。
[0059]
在varian liquid state nmr仪器上进行1h和
13
c nmr图谱测试。溶剂为cdcl3或dmso-d6。溶剂中如有内标四甲基硅烷,化学位移则参照四甲基硅烷(δ=0.00ppm);否则,若以cdcl3为溶剂,1h nmr图谱化学位移则参照残留溶剂(δ=7.26ppm);若以dmso-d6为溶剂,1h nmr图谱化学位移则参照残留溶剂h2o(δ=3.33ppm)。实施例中核磁数据使用下列缩写(或其组合)来解释1h nmr的多重性:s=单重,d=双重,t=三重,q=四重,p=五重,m=多重,br=宽。
[0060]
制备实施例
[0061]
实施例1:发光材料p-cz-bf2可按如下路线合成:
[0062][0063]
中间体p-cz-ome的合成:在100ml三口瓶中依次加入1-br(2.10g,7.95mmol,1.0当量)、咔唑(1.60g,9.54mmol,1.2当量)、叔丁醇钠(1.91g,19.88mmol,2.5当量)、pd2(dba)3(220mg,0.24mmol,0.03当量)和xphos(229mg,0.48mmol,0.06当量),然后抽换氮气三次,注射加入甲苯(35ml),在120℃油浴下反应70小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=20:1-5:1,得到黄棕色固体2.63g,收率94%。1h nmr(500mhz,cdcl3)δ3.90(s,3h),7.23(d,j=2.0hz,1h),7.28-7.37(m,4h),7.43-7.46(m,2h),7.53(d,j=8.5hz,2h),7.82-7.87(m,1h),7.97(dt,j=8.0,1.0hz,1h),8.04(d,j=8.0hz,1h),8.16(dt,j=8.0,1.0hz,2h),8.80(d,j=4.5hz,1h)。
[0064]
中间体p-cz-oh的合成:在干燥的250ml三口瓶中加入p-cz-ome(2.98g,8.51mmol,1.0当量),干燥的二氯甲烷(100ml)。然后在-15℃下滴加三溴化硼(1.61ml,17.03mmol,2.0当量),移至室温搅拌反应10小时。碳酸氢钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲
烷=20:1-5:1,得到黄色固体1.82g,收率为64%。1h nmr(500mhz,dmso-d6)δ7.17(d,j=2.0hz,1h),7.20(dd,j=8.5,2.0hz,1h),7.30-7.33(m,2h),7.45-7.54(m,5h),8.10(td,j=8.0,2.0hz,1h),8.26(d,j=7.7hz,2h),8.33(d,j=8.5hz,2h),8.70(dd,j=5.0,1.0hz,1h),14.58(s,1h)。
[0065]
发光材料p-cz-bf2的合成:在干燥的250ml三口瓶中加入p-cz-oh(1.82g,5.41mmol,1.0当量),二氯甲烷(100ml),然后在-15℃下加入三氟化硼乙醚(6.8ml,54.1mmol,10当量),移至室温搅拌反应10小时后再加入n,n-二异丙基乙胺(14.2ml,81.15mmol,15当量),继续反应11小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:3,得到黄绿色固体1.75g,收率为84%。1h nmr(500mhz,dmso-d6)δ7.32-7.35(m,2h),7.39(d,j=2.0hz,1h),7.43(dd,j=8.5,2.0hz,1h),7.47-7.50(m,2h),7.57(d,j=8.0hz,2h),7.90-7.93(m,1h),8.27(dt,j=8.0,1.0hz,2h),8.52(d,j=8.5hz,1h),8.54-8.58(m,1h),8.71(d,j=8.5hz,1h),8.83(d,j=6.0hz,1h)。
[0066]
实施例2:发光材料p-phcz-bf2可按如下路线合成:
[0067][0068]
中间体p-phcz-ome的合成:在100ml三口瓶中依次加入1-br(2.0g,7.57mmol,1.0当量)、苯基咔唑(2.66g,8.33mmol,1.1当量)、叔丁醇钠(1.82g,18.93mmol,2.5当量)、pd2(dba)3(208mg,0.23mmol,0.03当量)和xphos(217mg,0.45mmol,0.06当量),然后抽换氮气三次,注射加入甲苯(40ml),在125℃油浴下反应72小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=20:1-1:1,得到棕色固体2.76g,收率73%。1h nmr(400mhz,cdcl3)δ3.93(s,3h),7.28(d,j=2.0hz,1h),7.35
–
7.41(m,4h),7.50(t,j=7.6hz,4h),7.61(d,j=8.4hz,2h),7.71
–
7.77(m,6h),7.89(t,j=7.6hz,1h),8.00(d,j=8.0hz,1h),8.07(d,j=8.0hz,1h),8.42(d,j=1.6hz,2h),8.82(d,j=4.4hz,1h)。
[0069]
中间体p-phcz-oh的合成:在干燥的250ml三口瓶中加入p-phcz-ome(2.76g,5.49mmol,1.0当量),干燥的二氯甲烷(60ml)。然后在-15℃下滴加三溴化硼(1.04ml,10.98mmol,2.0当量),移至室温搅拌反应20小时。碳酸氢钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=20:1-1:1,得到黄色固体2.14g,收率为80%。1h nmr(500mhz,dmso-d6)δ7.24(d,j=2.0hz,1h)7.27(dd,j=8.5,2.0hz,1h),7.35
–
7.39(m,2h),7.49
–
7.53(m,5h),7.63(d,j=8.5hz,2h),7.82
–
7.85(m,6h),8.11(td,j=8.0,2.0hz,1h),8.35
–
8.38(m,2h),8.71(ddd,j=5.0,2.0,1.0hz,1h),8.76(d,j=1.5hz,2h),14.64(s,1h)。
[0070]
发光材料p-phcz-bf2的合成:在干燥的100ml三口瓶中加入p-phcz-oh(2.0g,4.09mmol,1.0当量),二氯甲烷(50ml),然后在-15℃下加入三氟化硼乙醚(5.2ml,40.93mmol,10当量),移至室温搅拌反应20小时后再加入n,n-二异丙基乙胺(10.72ml,61.35mmol,15当量),继续反应24小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:3,得到黄绿色固体1.94g,收率为88%。1h nmr(400mhz,chloroform-d)δ7.35
–
7.41(m,3h),7.48
–
7.54(m,5h),7.65
–
7.75(m,9h),8.09(d,j=8.4hz,1h),8.20
–
8.29(m,2h),8.40(s,2h),8.79(d,j=6.0hz,1h).
[0071]
实施例3:发光材料p-tbucz-bf2可按如下路线合成:
[0072][0073]
中间体p-tbucz-oh的合成:在50ml三口瓶中依次加入2-br(400mg,1.60mmol,1.0当量)、3,6-二叔丁基咔唑(581mg,2.08mmol,1.3当量)、叔丁醇钠(384mg,4.0mmol,2.5当量)、pd2(dba)3(44mg,0.048mmol,0.03当量)和xphos(92mg,0.19mmol,0.12当量),然后抽换氮气三次,注射加入甲苯(18ml),在120℃油浴下反应96小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=20:1-3:1,得到白色固体234mg,收率33%。1h nmr(500mhz,cdcl3)δ1.47(s,18h),7.14(dd,j=8.5,2.0hz,1h),7.27(d,j=2.0hz,1h),7.30(ddd,j=7.5,5.0,1.0hz,1h),7.48(dd,j=9.0,2.0hz,2h),7.53(d,j=8.5hz,2h),7.88-7.91(m,1h),7.97-8.00(m,1h),8.14(d,j=1.0hz,2h),8.56(ddd,j=5.0,2.0,1.0hz,1h),14.76(s,1h)。
[0074]
发光材料p-tbucz-bf2的合成:在干燥的50ml三口瓶中加入p-tbucz-oh(230mg,0.52mmol,1.0当量),二氯甲烷(15ml),然后在-15℃下加入三氟化硼乙醚(0.2ml,1.56mmol,3.0当量),移至室温搅拌反应2小时后再加入n,n-二异丙基乙胺(0.37ml,2.09mmol,4.0当量),继续反应12小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:
石油醚/二氯甲烷=5:1-1:2,得到黄绿色固体139mg,收率为54%。1h nmr(500mhz,cdcl3)δ1.47(s,18h),7.33(dd,j=8.5,2.0hz,1h),7.46(d,j=2.0hz,1h),7.48(dd,j=8.5,2.0hz,2h),7.54(d,j=8.5hz,2h),7.61-7.64(m,1h),8.02(d,j=8.5hz,1h),8.13(d,j=1.5hz,2h),8.18(d,j=8.5hz,1h),8.22-8.25(m,1h),8.76(d,j=5.5hz,1h)。
[0075]
实施例4:发光材料p-ptz-bf2可按如下路线合成:
[0076][0077]
中间体p-ptz-oh的合成:在50ml三口瓶中依次加入2-br(400mg,1.60mmol,1.0当量)、吩噻嗪(415mg,2.08mmol,1.3当量)、叔丁醇钠(461mg,4.8mmol,3.0当量)和醋酸钯(11mg,0.048mmol,0.03当量),然后抽换氮气三次,注射加入三叔丁基膦(260mg,0.13mmol,0.08当量,10wt%甲苯)和1,4二氧六环(15ml),在100℃油浴下反应50小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=50:1-10:1,得到棕黄色固体435mg,收率74%。1h nmr(500mhz,cdcl3)δ6.70(dd,j=8.0,1.5hz,2h),6.83(dd,j=8.5,2.5hz,1h),6.92(td,j=7.5,1.5hz,2h),6.97
–
7.01(m,3h),7.13(dd,j=7.5,1.5hz,2h),7.26
–
7.29(m,1h),7.84
–
7.92(m,3h),8.53(ddd,j=5.0,2.0,1.0hz,1h),14.63(s,1h)。
[0078]
发光材料p-ptz-bf2的合成:在干燥的50ml三口瓶中加入p-ptz-oh(400mg,1.09mmol,1.0当量),二氯甲烷(10ml),然后在-15℃下加入三氟化硼乙醚(0.4ml,3.26mmol,3.0当量),移至室温搅拌反应3小时后再加入n,n-二异丙基乙胺(0.76ml,4.36mmol,4.0当量),继续反应17小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:1,得到橙色固体260mg,收率为57%。1h nmr(500mhz,cdcl3)δ6.13(dd,j=8.0,1.5hz,2h),6.63(td,j=7.5,1.5hz,2h),6.69(td,j=7.5,1.5hz,2h),6.72(dd,j=7.5,1.5hz,2h),7.06(dd,j=8.5,2.0hz,1h),7.23(d,j=2.0hz,1h),7.67(ddd,j=7.5,6.0,1.0hz,1h),8.06(d,j=8.5hz,1h),8.18(d,j=8.5hz,1h),8.26(ddd,j=9.0,7.5,1.5hz,1h),8.78(d,j=6.0hz,1h)。
[0079]
实施例5:发光材料p-pxz-bf2可按如下路线合成:
[0080][0081]
中间体p-pxz-oh的合成:在50ml三口瓶中依次加入2-br(400mg,1.60mmol,1.0当
量)、吩噁嗪(381mg,2.08mmol,1.3当量)、叔丁醇钠(461mg,4.8mmol,3.0当量)和醋酸钯(11mg,0.048mmol,0.03当量),然后抽换氮气三次,注射加入三叔丁基膦(260mg,0.13mmol,0.08当量,10wt%甲苯)和1,4二氧六环(15ml),在100℃油浴下反应50小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=50:1-10:1,得到黄色固体570mg,收率88%。1h nmr(500mhz,cdcl3)δ6.13(dd,j=8.0,1.5hz,2h),6.63(ddd,j=8.0,7.0,1.5hz,2h),6.69(td,j=7.5,1.5hz,2h),6.73(dd,j=7.5,1.5hz,2h),7.06(dd,j=8.5,2.0hz,1h),7.23(d,j=2.0hz,1h),7.67(ddd,j=7.5,6.0,1.0hz,1h),8.06(d,j=8.5hz,1h),8.18(d,j=8.5hz,1h),8.26(ddd,j=9.0,7.5,1.5hz,1h),8.78(d,j=6.0hz,1h)。
[0082]
发光材料p-pxz-bf2的合成:在干燥的50ml三口瓶中加入p-pxz-oh(570mg,1.62mmol,1.0当量),二氯甲烷(12ml),然后在-15℃下加入三氟化硼乙醚(0.6ml,4.85mmol,3.0当量),移至室温搅拌反应2小时后再加入n,n-二异丙基乙胺(0.1.1ml,6.48mmol,4.0当量),继续反应17小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:1,得到橙色固体222mg,收率为39%。1h nmr(500mhz,cdcl3)δ6.71(dd,j=9.0,2.5hz,1h),6.84(d,j=2.5hz,1h),7.18(td,j=7.5,1.5hz,2h),7.30(td,j=7.5,1.5hz,2h),7.36(dd,j=8.0,1.5hz,2h),7.40
–
7.43(m,3h),7.64(d,j=9.0hz,1h),7.91(d,j=8.5hz,1h),8.05(ddd,j=8.5,7.5,1.5hz,1h),8.57(d,j=6.0hz,1h)。
[0083]
实施例6:发光材料p-dmac-bf2可按如下路线合成:
[0084][0085]
中间体p-dmac-ome的合成:在50ml三口瓶中依次加入1-br(1.0g,3.79mmol,1.0当量)、吖啶(951mg,4.54mmol,1.2当量)、叔丁醇钠(728mg,7.58mmol,2.0当量)、pd2(dba)3(104mg,0.11mmol,0.03当量)和s-phos(93mg,0.23mmol,0.06当量),然后抽换氮气三次,注射加入甲苯(15ml),在110℃油浴下反应67小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋
洗剂:石油醚/乙酸乙酯=50:1-10:1,得到棕黄色固体1.35g,收率91%。1h nmr(500mhz,cdcl3)δ1.72(s,6h),3.83(s,3h),6.42(dd,j=8.0,1.5hz,2h),6.93
–
6.96(m,3h),7.00(ddd,j=8.5,7.0,1.5hz,2h),7.08(dd,j=8.0,1.5hz,1h),7.26
–
7.29(m,1h),7.47(dd,j=7.5,1.5hz,2h),7.78(td,j=7.5,2.0hz,1h),7.92(dt,j=8.0,1.0hz,1h),8.02(d,j=8.0hz,1h),8.76(ddd,j=5.0,2.0,1.0hz,1h)。
[0086]
中间体p-dmac-oh的合成:在干燥的50ml三口瓶中加入p-dmac-ome(1.18g,3.0mmol,1.0当量),干燥的二氯甲烷(20ml)。然后在-15℃下滴加三溴化硼(0.85ml,9.0mmol,3.0当量),移至室温搅拌反应19小时。碳酸氢钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=20:1-1:1,得到黄色固体760mg,收率为67%。1h nmr(500mhz,cdcl3)δ1.69(s,6h),6.48(dd,j=8.0,1.5hz,2h),6.88(dd,j=8.5,2.0hz,1h),6.93(td,j=7.5,1.5hz,2h),6.97
–
7.01(m,2h),7.05(d,j=2.0hz,1h),7.32(ddd,j=7.5,5.0,1.0hz,1h),7.46(dd,j=7.5,1.5hz,2h),7.91(td,j=8.0,2.0hz,1h),7.98
–
8.01(m,1h),8.04(d,j=8.5hz,1h),8.58(ddd,j=5.0,2.0,1.0hz,1h),14.67(s,1h)。
[0087]
发光材料p-dmac-bf2的合成:在干燥的50ml三口瓶中加入p-dmac-oh(620mg,1.64mmol,1.0当量),二氯甲烷(10ml),然后在-15℃下加入三氟化硼乙醚(2.07ml,10.4mmol,10当量),移至室温搅拌反应3小时后再加入n,n-二异丙基乙胺(4.3ml,24.6mmol,15当量),继续反应42小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:1,得到黄色固体570mg,收率为82%。1h nmr(400mhz,cdcl3)δ1.67(s,6h),6.55(d,j=7.6hz,2h),6.96
–
7.05(m,5h),7.24(s,1h),7.47(d,j=7.2hz,2h),7.64(t,j=6.4hz,1h),8.05(d,j=8.4hz,1h),8.18(d,j=8.4hz,1h),8.24(t,j=8.0hz,1h),8.76(d,j=6.0hz,1h)。
[0088]
实施例7:发光材料p-dpa-bf2可按如下路线合成:
[0089][0090]
中间体p-dpa-ome的合成:在50ml三口瓶中依次加入1-br(788mg,2.98mmol,1.0当
量)、二苯胺(606mg,3.58mmol,1.2当量)、叔丁醇钠(573mg,5.96mmol,2.0当量)、pd2(dba)3(82mg,0.09mmol,0.03当量)和s-phos(73mg,0.18mmol,0.06当量),然后抽换氮气三次,注射加入甲苯(12ml),在110℃油浴下反应96小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=50:1-10:1,得到棕黄色固体975mg,收率93%。1h nmr(400mhz,cdcl3)δ3.70(s,3h),6.68(s,1h),6.74(d,j=8.8hz,1h),7.07(t,j=7.2hz,2h),7.16(d,j=8.0hz,4h),7.21(d,j=6.4hz,1h),7.26
–
7.31(m,4h),7.69(d,j=8.4hz,1h),7.76(t,j=8.0hz,1h),7.86(d,j=8.0hz,1h),8.72(d,j=4.8hz,1h)。
[0091]
中间体p-dpa-oh的合成:在干燥的50ml三口瓶中加入p-dpa-ome(925mg,2.62mmol,1.0当量),干燥的二氯甲烷(30ml)。然后在-15℃下滴加三溴化硼(0.74ml,7.87mmol,3.0当量),移至室温搅拌反应26小时。碳酸氢钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=20:1-1:1,得到白色固体297mg,收率为33%。1h nmr(400mhz,cdcl3)δ6.57(dd,j=8.4,2.4hz,1h),6.69(s,1h),7.09(t,j=7.2hz,2h),7.15
–
7.22(m,5h),7.27
–
7.31(m,4h),7.60(d,j=8.4hz,1h),7.79
–
7.86(m,2h),8.46(d,j=4.8hz,1h),14.40(s,1h)。
[0092]
发光材料p-dpa-bf2的合成:在干燥的50ml三口瓶中加入p-dpa-oh(250mg,0.74mmol,1.0当量),二氯甲烷(10ml),然后在-15℃下加入三氟化硼乙醚(0.93ml,7.39mmol,10当量),移至室温搅拌反应1.5小时后再加入n,n-二异丙基乙胺(1.94ml,11.1mmol,15当量),继续反应26小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:1,得到黄色固体245mg,收率为86%。1h nmr(400mhz,cdcl3)δ6.62
–
6.66(m,2h),7.14
–
7.20(m,6h),7.31
–
7.35(m,4h),7.39(t,j=6.8hz,1h),7.59(d,j=8.8hz,1h),7.91(d,j=8.8hz,1h),8.04(t,j=8.0hz,1h),8.56(d,j=6.0hz,1h)。
[0093]
实施例8:发光材料m-cz-bf2可按如下路线合成:
[0094]
[0095]
中间体1-cl的合成:在250ml三口瓶中依次加入2-溴吡啶(13.53g,85.84mmol,2.0当量)、5-氯-2-甲氧基苯硼酸(8.0g,42.92mmol,1.0当量)、碳酸铯(34.96g,107.3mmol,2.5当量)和四三苯基膦钯(992mg,0.86mmol,0.02当量),然后抽换氮气三次,注射加入乙醇(120ml)和水(50ml),在80℃油浴下反应96小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=50:1-10:1,得到无色液体7.07g,收率75%。1h nmr(500mhz,cdcl3)δ3.86(s,3h),6.94(d,j=8.5hz,1h),7.27
–
7.29(m,1h),7.33(dd,j=8.5,2.5hz,1h),7.75
–
7.78(m,2h),7.83(dt,j=7.5,1.0hz,1h),8.73(d,j=4.5hz,1h)。
[0096]
中间体m-cz-ome的合成:在100ml三口瓶中依次加入1-cl(2.0g,9.10mmol,1.0当量)、咔唑(1.83g,10.93mmol,1.2当量)、叔丁醇钠(2.19g,22.75mmol,2.5当量)、pd2(dba)3(250mg,0.27mmol,0.03当量)和xphos(260mg,0.55mmol,0.06当量),然后抽换氮气三次,注射加入邻二甲苯(30ml),在140℃油浴下反应50小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=30:1-1:1,得到棕色固体2.93g,收率92%。1h nmr(500mhz,cdcl3)δ4.01(s,3h),7.23
–
7.26(m,1h),7.26
–
7.34(m,3h),7.38
–
7.42(m,4h),7.57(dd,j=9.0,2.5hz,1h),7.84(t,j=7.5hz,1h),7.93(d,j=8.0hz,1h),7.96(d,j=2.5hz,1h),8.14(dt,j=7.5,1.0hz,2h),8.73(d,j=2.0hz,1h)。
[0097]
中间体m-cz-oh的合成:在干燥的500ml三口瓶中加入m-cz-ome(2.94g,8.39mmol,1.0当量),干燥的二氯甲烷(150ml)。然后在-15℃下滴加三溴化硼(1.59ml,16.78mmol,2.0当量),移至室温搅拌反应18小时。碳酸氢钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/乙酸乙酯=20:1-10:1,得到黄色固体2.25g,收率为80%。1h nmr(500mhz,cdcl3)δ7.27
–
7.37(m,5h),7.40
–
7.44(m,3h),7.47(dd,j=8.5,2.5hz,1h),7.83
–
7.86(m,2h),7.94(d,j=2.5hz,1h),8.17(d,j=7.5hz,2h),8.61(d,j=5.0hz,1h),14.39(s,1h)。
[0098]
发光材料m-cz-bf2的合成:在干燥的250ml三口瓶中加入m-cz-oh(2.25g,6.69mmol,1.0当量),二氯甲烷(100ml),然后在-15℃下加入三氟化硼乙醚(2.53ml,20.07mmol,3.0当量),移至室温搅拌反应9小时后再加入n,n-二异丙基乙胺(4.67ml,26.76mmol,4.0当量),继续反应28小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:1,得到黄色固体970mg,收率为38%。1h nmr(500mhz,cdcl3)δ7.30
–
7.35(m,4h),7.41
–
7.45(m,3h),7.66
–
7.69(m,2h),8.02(d,j=2.5hz,1h),8.07(d,j=8.5hz,1h),8.17(dt,j=7.5,1.0hz,2h),8.21(ddd,j=9.0,7.5,1.5hz,1h),8.81(dd,j=6.0,1.5hz,1h)。
[0099]
实施例9:发光材料m-phcz-bf2可按如下路线合成:
[0100][0101]
中间体m-phcz-ome的合成:在100ml三口瓶中依次加入1-cl(2.0g,9.10mmol,1.0当量)、苯基咔唑(3.49g,10.93mmol,1.2当量)、叔丁醇钠(2.19g,22.75mmol,2.5当量)、pd2(dba)3(250mg,0.27mmol,0.03当量)和xphos(260mg,0.55mmol,0.06当量),然后抽换氮气三次,注射加入邻二甲苯(30ml),在140℃油浴下反应48小时。反应冷却至室温,加水淬灭,乙酸乙酯萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=5:1-1:100,得到棕色固体2.98g,收率65%。1h nmr(500mhz,cdcl3)δ4.03(s,3h),7.27(d,j=9.0hz,1h),7.33
–
7.37(m,3h),7.47
–
7.51(m,6h),7.64(dd,j=8.5,3.0hz,1h),7.68(dd,j=8.5,2.0hz,2h),7.72
–
7.75(m,4h),7.89
–
7.90(m,1h),7.96(d,j=8.0hz,1h),8.01(d,j=2.5hz,1h),8.40(d,j=2.0hz,2h),8.78(d,j=2.0hz,1h)。
[0102]
中间体m-phcz-oh的合成:在干燥的500ml三口瓶中加入m-phcz-ome(2.98g,5.93mmol,1.0当量),干燥的二氯甲烷(150ml)。然后在-15℃下滴加三溴化硼(1.12ml,11.86mmol,2.0当量),移至室温搅拌反应18小时。碳酸氢钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=20:1-1:1,得到黄色固体2.48g,收率为86%。1h nmr(500mhz,dmso-d6)δ7.25(d,j=8.7hz,1h),7.34
–
7.38(m,2h),7.43(d,j=8.5hz,2h),7.49
–
7.53(m,5h),7.58(dd,j=8.5,2.5hz,1h),7.79(dd,j=8.5,2.0hz,2h),7.81
–
7.84(m,4h),8.02(td,j=8.5,2.0hz,1h),8.34
–
8.36(m,2h),8.70
–
8.72(m,1h),8.74(d,j=2.0hz,2h),14.49(s,1h)。
[0103]
发光材料m-phcz-bf2的合成:在干燥的250ml三口瓶中加入m-phcz-oh(2.28g,4.67mmol,1.0当量),二氯甲烷(50ml),然后在-15℃下加入三氟化硼乙醚(5.89ml,46.67mmol,10当量),移至室温搅拌反应9小时后再加入n,n-二异丙基乙胺(12.26ml,70.05mmol,15当量),继续反应28小时。反应完毕后用碳酸钠溶液淬灭,二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,粗品用硅胶柱色谱分离,淋洗剂:石油醚/二氯甲烷=20:1-1:1,得到黄色固体1.70g,收率为68%。1h nmr(500mhz,cdcl3)δ
7.35
–
7.39(m,2h),7.42(dd,j=8.5,1.0hz,2h),7.47
–
7.51(m,5h),7.68
–
7.71(m,3h),7.73
–
7.75(m,5h),8.07(d,j=2.5hz,1h),8.11(d,j=8.5hz,1h),8.23(ddd,j=9.0,7.5,1.5hz,1h),8.43(dd,j=1.8,0.5hz,2h),8.83(d,j=6.0hz,1h)。
[0104]
性能评价实施例
[0105]
以下对本发明上述实施例中所制备的配合物进行光物理、电化学和热重分析:
[0106]
光物理分析:磷光发射光谱、荧光发射光谱、三线态寿命、激发态寿命均在horiba fl3-11光谱仪上测试完成。测试条件:低温和室温发射光谱中,所有样品均为甲苯(色谱级)稀溶液(10-5-10-6
m);发光量子效率(plqy)和发光衰减曲线均为发光材料10wt%掺杂的depeo薄膜样品测得。
[0107]
图1分别是发光材料p-cz-bf2、p-tbucz-bf2、p-phcz-bf2、p-dmac-bf2、p-dpa-bf2、p-ptz-bf2、p-pxz-bf2、m-cz-bf2和m-phcz-bf2的甲苯溶液在室温下的发射光谱谱图。
[0108]
图2是发光材料p-cz-bf2、p-tbucz-bf2、p-phcz-bf2、p-dmac-bf2、p-dpa-bf2、p-ptz-bf2、p-pxz-bf2、m-cz-bf2和m-phcz-bf2的depeo薄膜在室温下的发射光谱谱图。
[0109]
图3是发光材料p-cz-bf2、p-tbucz-bf2、p-phcz-bf2、p-dmac-bf2、p-dpa-bf2、p-ptz-bf2、p-pxz-bf2、m-cz-bf2和m-phcz-bf2的depeo薄膜发光衰减(归一化发光强度-时间)曲线。
[0110]
表1.发光材料的光物理性质
[0111]
发光材料peak/nmplqy/%p-cz-bf243899p-tbucz-bf245999p-phcz-bf245599p-dpa-bf247493p-pxz-bf256524p-ptz-bf2457、58336p-dmac-bf252878m-cz-bf248799m-phcz-bf249199
[0112]
注:peak是指发光材料室温下在甲苯溶液中发射光谱的最强发射峰。plqy是指发光材料10wt%掺杂的depeo薄膜样品绝对发光量子效率。
[0113]
从附图1、附图2和表1可知:其一,材料发光颜色容易调节:在保持受体结构不变的情况下,只是通过简单的调节给体的结构即可是使材料的发光颜色覆盖整个蓝光至橙光的可见光区;其二,材料量子效率高:材料有很高的发光量子效率(plqy),尤其是蓝光材料可高达99%。其三,热致延迟荧光材料:从附图3材料的depeo薄膜发光衰减(归一化发光强度-时间)曲线可知,其大部分都是双指数衰减方式,为典型的热致延迟荧光材料,理论上均可通过反向系间窜跃充分利用处于三线态激子,达到100%的量子效率。以上这些性质均有利于其作为掺杂发光体在oled器件中的应用,并为解决目前急缺的蓝光发光材料提供一个有效的途径,以大大促进此领域的发展。
[0114]
器件实例
[0115]
所有材料在使用之前均经过高真空(10-5-10-6
torr)下梯度加热升华纯化。器件所
使用的铟锡氧化物(ito)基板均经过在去离子水、丙酮和异丙醇中依次进行超声处理。器件通过在真空度小于10-7
torr的压力下真空热蒸镀制备。阳极电极为厚度为铟锡氧化物(ito),阴极由厚度为的li2co3和的al组成。所有器件制备完毕后在氮气手套箱中玻璃盖和环氧树脂封装,并在包装内加入吸湿剂。
[0116]
发光材料p-cz-bf2和m-phcz-bf2作为发光体在不同主体材料和传输材料下的器件结构如下所示:
[0117]
ito/hatcn(10nm)/tapc(60nm)/mcbp:ppt:p-cz-bf2(1:1,5%,30nm)/ppt(2nm)/bepp2:li2co3(5%,35nm)/li2co3(1nm)/al,器件1;
[0118]
ito/hatcn(10nm)/tapc(60nm)/mcbp:ppt:p-cz-bf2(1:2,5%,30nm)/ppt(2nm)/bepp2:li2co3(5%,35nm)/li2co3(1nm)/al,器件2;
[0119]
ito/hatcn(10nm)/tapc(60nm)/mcbp:ppt:p-cz-bf2(1:1,5%,30nm)/ppt(2nm)/ppt:li2co3(5%,35nm)/li2co3(1nm)/al,器件3;
[0120]
ito/hatcn(10nm)/tapc(60nm)/mcbp:ppt:p-cz-bf2(1:2,5%,30nm)/ppt(2nm)/ppt:li2co3(5%,35nm)/li2co3(1nm)/al,器件4;
[0121]
ito/hatcn(10nm)/npb(40nm)/mcbp(10nm)/ppt:m-phcz-bf2(10%,30nm)/ppt(5nm)/bepp2:li2co3(5%,30nm)/li2co3(1nm)/al,器件5;
[0122]
ito/hatcn(10nm)/npb(50nm)/ppt:m-phcz-bf2(10%,30nm)/ppt(5nm)/bepp2:li2co3(5%,30nm)/li2co3(1nm)/al,器件6;
[0123]
ito/hatcn(10nm)/npb(40nm)/ppt:m-phcz-bf2(10%,30nm)/ppt(5nm)/bepp2:li2co3(5%,30nm)/li2co3(1nm)/al,器件7;
[0124]
ito/hatcn(15nm)/npb(50nm)/ppt:m-phcz-bf2(10%,30nm)/ppt(5nm)/bepp2:li2co3(5%,30nm)/li2co3(1nm)/al,器件8;
[0125]
上述器件所使用材料的分子结构如下所示:
[0126][0127]
图4是发光材料p-cz-bf2作为发光体在双主体材料不同比例及电子传输材料不同掺杂的器件电致发光谱图。
[0128]
图5是发光材料p-cz-bf2作为发光体在双主体材料不同比例及电子传输材料不同掺杂的器件电流密度-电压-发光强度曲线。
[0129]
图6是发光材料p-cz-bf2作为发光体在双主体材料不同比例及电子传输材料不同掺杂的器件外部量子效率-电流密度曲线。
[0130]
图7是发光材料m-phcz-bf2作为发光体在不同功能层厚度下的器件电致发光谱图。
[0131]
图8是发光材料m-phcz-bf2作为发光体在不同功能层厚度下的器件电流密度-电压-发光强度曲线。
[0132]
图9是发光材料m-phcz-bf2作为发光体在不同功能层厚度下的器件外部量子效率-电流密度曲线。
[0133]
从附图8、9中可以看到,以发光材料m-phcz-bf2作为发光体在不同器件下具有良好的器件性能,器件5、6、7、8最大外部量子效率分别高达23.9%、25.6%、22.4%、10.2%;器件5、6、7、8最大发光亮度分别高达17493、12633、14029和13250cd/m2。器件数据充分表明此类含有酚氧-吡啶螯合二氟化硼受体的有机发光材料作为发光材料是可行的,其优异的性能可以作为热致延迟荧光材料,其在oled领域有着巨大的应用前景,并促进此领域的进一步发展。
[0134]
本领域的普通技术人员可以理解,上述各实施方式是实现本发明的具体实施例,而在实际应用中,可以在形式上和细节上对其作各种改变,而不偏离本发明的精神和范围。例如,在不背离本发明的精神的情况下,这里描述的许多材料和结构可以用其它材料和结
构代替。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
