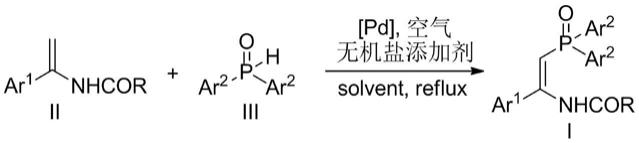
1.本发明提供一种低成本的关键中间体3-{3-氟-2-[3-(2-甲氧基-5-(三氟甲基)苯基)脲基]苯基}丙烯酸甲酯的制备方法,属于医药化工技术领域。
背景技术:
[0002]
莱特莫韦(letermovir),化学名:(4s)-2-{8-氟-2-[4-(3-甲氧基苯基)-1-哌嗪基]-3-[2-甲 氧基-5-(三氟甲基)苯基]-3,4二氢)-4-喹唑啉基}乙酸,是由德国aicurisanti
‑ꢀ
infectivecuresgmbh公司首先研发,然后由美国默沙东制药公司(merck&co)收购并申报上市 的一种用于预防与治疗术后成人巨细胞病毒的药物,其结构式如下式i目前制备莱特莫韦主要有以下三种方法:
①
wo2013127970专利文献中以邻氟苯胺为起始原料,经过成脲,醋酸钯催化偶联,氯 代,与侧链对接,拆分、水解等步骤,最终得到莱特莫韦。
[0003]
②
us2009221822专利文献中以2-氟-6-溴苯胺为起始原料,同样经过成脲,钯催化 偶联,氯代,与侧链对接,拆分、水解等步骤,最终得到莱特莫韦。
[0004]
③
wo2015088931专利文献中2-氟-6-溴苯胺为起始原料,经过钯催化偶联,氨基保 护,2-甲氧基-5-三氟甲基苯胺取代,氯代,与侧链对接,环化,拆分、水解等步骤,最终得 到莱特莫韦。
[0005]
以上三条路线无论是选择哪一条,均要使用钯催化剂催化合成母核(式ii)或其衍生 物。
[0006]
钯催化剂价格昂贵,其使用直接导致现有产品制备成本大幅度提升。
技术实现要素:
[0007]
为了降低产品的制备成本,本发明设计了一条莱特莫韦关键中间体3-(3-氟-2-(3-(2-甲氧基-5-(三氟甲基)苯基)脲基)苯基)丙烯酸甲酯(式vi)合成路线,然后化合物vi再以乙酸异丙酯为溶剂,dbu做碱制备得到化合物vii,或进一步采用专利wo2015088931的方法得到化合物viii,避开了钯催化剂的应用,大幅度的降低了产品制备成本。
[0008]
本发明提供一种低成本的关键中间体3-(3-氟-2-(3-(2-甲氧基-5-(三氟甲基)苯基)脲基)苯基)丙烯酸甲酯的制备方法,其操作简单、收率高、成本低,特别适用于工业化生产。
[0009]
本发明的技术方案是一种莱特莫韦关键中间体3-(3-氟-2-(3-(2-甲氧基-5-(三氟甲基)苯基)脲基)苯基)丙烯酸甲酯的制备方法,包括以下步骤:(1)以2-氟-6-溴苯胺为起始原料,敷酸剂条件下与酰氯反应得到化合物ii;(2)碱性条件下,化合物ii与正丁基锂反应,然后再与dmf中反应得到化合物iii;(3)化合物iii与二甲基膦酰基乙酸甲酯在碱性条件下反应得到化合物iv;(4)化合物iv在乙醇浓盐酸混合溶剂中回流得到化合物v;(5)以2-甲氧基-5-三氟甲基苯胺为原料,先与三光气反应制备异氰酸酯中间体,再与化合物v反应得到化合物3-{3-氟-2-[3-(2-甲氧基-5-(三氟甲基)苯基)脲基]苯基}丙烯酸甲酯(vi)。
[0010]
所述步骤(1)中的酰氯为特戊酰氯、异戊酰氯,乙酰氯。
[0011]
所述步骤(1)中的敷酸剂为磷酸氢二钠、碳酸氢钠、碳酸钠、碳酸钾。
[0012]
所述步骤(2)中的碱性条件为tmeda、dipa,其比例约为16:1~16:5。
[0013]
所述步骤(3)中的强碱为氢化钠、hmdsli、hmdsk、lda。
[0014]
所述步骤(4)中的乙醇浓盐酸混合溶剂的体积比为1:1~8:3。
[0015]
所述步骤(5)中产品制备时所述第二溶剂选自由四氢呋喃、二氧六环、2-甲基四氢呋喃、甲苯组成的组中的一种或多种。
[0016]
具体实施例:1、n-(2-溴-6-氟苯基)烷基酰胺制备1)将49.2克(0.26mol)2-溴-6-氟苯胺加入至492毫升乙酸异丙酯,180毫升水的混合溶剂中,然后加入49.2克磷酸氢二钠,搅拌,然后将42.2克(0.35mmol)特戊酰氯滴加至体系中,反应体系在室温下反应3小时,反应完毕后,分液,取有机相,有机相再用240ml 1m的盐酸水溶液洗涤一次,240毫升饱和食盐水溶液洗涤一次,有机相浓缩,剩余物再正己烷中打浆得到白色固体,过滤,真空干燥得到61.4克固体,收率86.15%。
[0017]
2)将38.4克(0.20mol)2-溴-6-氟苯胺加入至384毫升乙酸异丙酯,140毫升水的混合溶剂中,然后加入38.4克磷酸氢二钠,搅拌,然后将32.9克(0.27mmol)异戊酰氯滴加至体系中,反应体系在室温下反应3小时,反应完毕后,分液,取有机相,有机相再用187ml 1m的盐酸水溶液洗涤一次,187毫升饱和食盐水溶液洗涤一次,有机相浓缩,剩余物再正己烷中打浆得到白色固体,过滤,真空干燥得到46.7克固体,收率84.34%。
[0018]
3)将442.8克(2.34mol)2-溴-6-氟苯胺加入至4428毫升乙酸异丙酯,1620毫升水的混合溶剂中,然后加入443.1克磷酸氢二钠,搅拌,然后将247.5克(3.15mmol)乙酰氯滴加至体系中,反应体系在室温下反应3小时,反应完毕后,分液,取有机相,有机相再用2160ml 1m的盐酸水溶液洗涤一次,2160毫升饱和食盐水溶液洗涤一次,有机相浓缩,剩余物再正己烷中打浆得到白色固体,过滤,真空干燥得到467.8克固体,收率72.9%。
[0019]
4)将738克(3.9mol)2-溴-6-氟苯胺加入至7380毫升乙酸异丙酯,2700毫升水的混合溶剂中,然后加入655.1克碳酸氢钠,搅拌,然后将633克(5.25mmol)特戊酰氯滴加至体系中,反应体系在室温下反应3小时,反应完毕后,分液,取有机相,有机相再用3600ml 1m的盐酸水溶液洗涤一次,3600毫升饱和食盐水溶液洗涤一次,有机相浓缩,剩余物再正己烷中打浆得到白色固体,过滤,真空干燥得到910.4克固体,收率85.16%。
[0020]
5)将1.31千克(5.98mol)2-溴-6-氟苯胺加入至11.3升乙酸异丙酯,4.14升水的混合溶剂中,然后加入634.8克碳酸钠,搅拌,然后将970.6克(8.05mmol)特戊酰氯滴加至体系中,反应体系在室温下反应3小时,反应完毕后,分液,取有机相,有机相再用5.5升 1m的盐酸水溶液洗涤一次,5.5升饱和食盐水溶液洗涤一次,有机相浓缩,剩余物再正己烷中打浆得到白色固体,过滤,真空干燥得到1.34千克固体,收率81.74%。
[0021]
2、n-(2-氟-6-甲醛基苯基)特戊酰胺制备1)向250ml三口瓶中,依次加入27.4克 (0.1mol) n-(2-溴-6-氟苯基)特戊酰胺,12.8克tmeda(0.11mol), 3.44克dipa(6.8mmol),50g无水四氢呋喃,反应体系抽真空充氮循环三次,氮气保护,体系降温至-70~-78℃,在-70~-78℃下缓慢滴44毫升n-buli(2.5m,0.11mol)溶液,体系有升温现象,滴加完毕后,反应体系在此温度条件下继续反应2hrs;然后慢慢加入10.9克dmf(0.15mol),滴加完毕后,反应体系继续在-70~-78℃下反应1.5h,然后去除低温浴,使反应体系慢慢升到室温,反应体系由低温时的澄清慢慢变为室温时的白色浑浊状,向体系中加入16毫升水,淬灭反应,再加入32毫升乙酸乙酯,搅拌,有固体产生,用浓盐酸调节水相ph值约为3,盐酸用量约50克,固体溶解,呈黄色澄清状,分相,取有机相,有机相再用水萃洗两次,有机相浓缩,得黄色油状物18.1克,粗收率81.1%。
[0022]
2)向3l三口瓶中,依次加入295.9克 (1.08mol) n-(2-溴-6-氟苯基)烷基酰胺,138.24克tmeda(1.19mol), 111.4克dipa(0.22mol),540g无水四氢呋喃,反应体系抽真空充氮循环三次,氮气保护,体系降温至-70~-78℃,在-70~-78℃下缓慢滴475.2毫升n-buli(2.5m,1.19mol)溶液,体系有升温现象,滴加完毕后,反应体系在此温度条件下继续反应2hrs;然后慢慢加入117.7克dmf(1.62mol),滴加完毕后,反应体系继续在-70~-78℃下反应1.5h,然后去除低温浴,使反应体系慢慢升到室温,反应体系由低温时的澄清慢慢变为室温时的白色浑浊状,向体系中加入172.8毫升水,淬灭反应,再加入346毫升乙酸乙酯,搅拌,有固体产生,用浓盐酸调节水相ph值约为3,盐酸用量约540克,固体溶解,呈黄色澄清状,分相,取有机相,有机相再用水萃洗两次,有机相浓缩,得黄色油状物201.4克,粗收率
83.53%。
[0023]
3)向3l三口瓶中,依次加入328.8克 (1.2mol) n-(2-溴-6-氟苯基)烷基酰胺,153.6克tmeda(1.32mol), 206.4克dipa(0.408mol),600g无水四氢呋喃,反应体系抽真空充氮循环三次,氮气保护,体系降温至-70~-78℃,在-70~-78℃下缓慢滴528毫升n-buli(2.5m,1.32mol)溶液,体系有升温现象,滴加完毕后,反应体系在此温度条件下继续反应2hrs;然后慢慢加入130.8克dmf(1.8mol),滴加完毕后,反应体系继续在-70~-78℃下反应1.5h,然后去除低温浴,使反应体系慢慢升到室温,反应体系由低温时的澄清慢慢变为室温时的白色浑浊状,向体系中加入192毫升水,淬灭反应,再加入384毫升乙酸乙酯,搅拌,有固体产生,用浓盐酸调节水相ph值约为3,盐酸用量约600克,固体溶解,呈黄色澄清状,分相,取有机相,有机相再用水萃洗两次,有机相浓缩,得黄色油状物224.3克,粗收率83.7%。
[0024]
3、3-(3-氟-2-特戊酰胺基苯基)丙烯酸甲酯制备1)向300毫升四氢呋喃中加入5.76克钠氢(60%, 144 mmol),搅拌,然后将26.3克二甲基膦酰基乙酸甲酯(144mmol)滴加至上述体系中,滴完后,在室温搅拌1小时,将n-(2-氟-6-甲醛基苯基)特戊酰胺16克(71.7mmol)溶解在30毫升四氢呋喃中,然后加入至上述反应体系中,室温搅拌18小时,反应体系用300毫升水淬灭,用300毫升乙酸乙酯萃洗,分液,收集有机相,减压浓缩,得12.85克,粗收率为63.9%,产物不用进行进一步处理,直接用于下一步。
[0025]
2)向3升反应瓶中加入1.44升hmdsli四氢呋喃溶液(1mol/毫升四氢呋喃溶液,1.44mol),搅拌,然后将262克二甲基膦酰基乙酸甲酯(1.44mol)滴加至上述体系中,滴完后,在室温搅拌1小时,将n-(2-氟-6-甲醛基苯基)特戊酰胺160克(0.72mol)溶解在300毫升四氢呋喃中,然后加入至上述反应体系中,室温搅拌18小时,反应体系用3升水淬灭,用3升乙酸乙酯萃洗,分液,收集有机相,减压浓缩,得131.5克,粗收率65.39%,产物不用进行进一步处理,直接用于下一步。
[0026]
4、3-(2-胺基-3-氟苯基)丙烯酸甲酯制备1)将8.4克(0.03mmol)步骤3)所得产物加入至80毫升乙醇与80毫升浓盐酸的混合溶液中,室温搅拌4小时,然后加热至回流,并在回流温度下反应3小时,tlc跟踪监测,反应完毕后,冷却反应体系,在0-10℃搅拌3小时,过滤,所得白色滤饼用适量冷乙醇洗涤,干燥得4.8克产品,粗收率为81.97%。
[0027]
2)将134.4克(0.48mmol)步骤3)所得产物加入至1.28升乙醇与800毫升浓盐酸的混合溶液中,室温搅拌4小时,然后加热至回流,并在回流温度下反应3小时,tlc跟踪监测,反应完毕后,冷却反应体系,在0-10℃搅拌3小时,过滤,所得白色滤饼用适量冷乙醇洗涤,干燥得77.2克产品,粗收率为82.40%。
[0028]
3)将126克(0.45mmol)步骤3)所得产物加入至1.2升乙醇与450毫升浓盐酸的混合溶液中,室温搅拌4小时,然后加热至回流,并在回流温度下反应3小时,tlc跟踪监测,反应完毕后,冷却反应体系,在0-10℃搅拌3小时,过滤,所得白色滤饼用适量冷乙醇洗涤,干燥得68.2克产品,粗收率为77.65%。
[0029]
5、3-{3-氟-2-[3-(2-甲氧基-5-(三氟甲基)苯基)脲基]苯基}丙烯酸甲酯制备1)三光气加入10ml甲苯中,搅拌,冰水降温,三光气溶解。2-甲氧基-5-三氟甲基苯胺4g用10ml甲苯溶解后滴加到三光气溶液中,滴加过程中出现白色固体,反应液越来越粘
稠。滴加完毕,逐渐升温至回流,有盐酸和光气气体释放,白色固体逐渐溶解。保持回流8h以上(冷凝器最好加干燥管,避免水汽进入),反应液澄清。降温,减压蒸馏,蒸尽甲苯得到浅黄色液体为异氰酸酯(文献是蒸至出现固体),向残液中加入10ml乙腈,冰水降温。把步骤4所得产品取代苯胺4.0g加10ml乙腈稀释,滴加到异氰酸酯溶液中;滴加后,逐渐升温至回流,保持回流4h以上,析出较多白色固体。降温至室温,搅拌1h, 过滤,固体用少量乙腈洗涤一次,得到类白色结晶性粉末。在鼓风烘箱中50℃烘干一夜得到类白色固体约6g,粗收率为71.01%。
[0030]
2)三光气加入120ml甲苯中,搅拌,冰水降温,三光气溶解。2-甲氧基-5-三氟甲基苯胺48g用120ml甲苯溶解后滴加到三光气溶液中,滴加过程中出现白色固体,反应液越来越粘稠。滴加完毕,逐渐升温至回流,有盐酸和光气气体释放,白色固体逐渐溶解。保持回流8h以上(冷凝器最好加干燥管,避免水汽进入),反应液澄清。降温,减压蒸馏,蒸尽甲苯得到浅黄色液体为异氰酸酯(文献是蒸至出现固体),向残液中加入120ml四氢呋喃,冰水降温。把步骤4所得产品取代苯胺49.2g加100ml四氢呋喃稀释,滴加到异氰酸酯溶液中;滴加后,逐渐升温至回流,保持回流4h以上,析出较多白色固体。降温至室温,搅拌1h, 过滤,固体用少量四氢呋喃洗涤一次,得到类白色结晶性粉末。在鼓风烘箱中50℃烘干一夜得到类白色固体约68.4g,粗收率为65.81%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。