含有ras/raf蛋白干扰基团的hdac抑制剂及其制备方法
技术领域
1.本发明具体涉及利用以ras/raf为靶点的2,4,6-三甲氧基苯基-4-甲氧基-3-氨基苄基砜结构单元为帽基基团,以组蛋白去乙酰化酶(histone deacetylases,hdac)为靶点的羟肟酸或苯甲酰胺结构单元为锌离子结合基团,通过线性基团连接,得到一类含有ras/raf蛋白干扰基团的hdac抑制剂,以期获得高效低毒的抗肿瘤药物;本发明属于药物化学中这类化合物的制备方法以及它们在制备抗肿瘤药物方面的应用领域。
背景技术:
2.作为鸟嘌呤核苷酸结合蛋白的成员,ras蛋白及相关蛋白在众多的细胞调节过程中起着关键作用,如细胞增殖与分化、胞内胞液间运输、胞内氧化酶的产生、细胞骨架的调控和胞外信息的跨膜传递等。ras在细胞外信号的帮助下,作为调节gdp/gtp转化的分子开关,可导致包括raf-mek-erk通路在内的几个下游级联的激活。在细胞外刺激时,ras利用其固有的gtpase功能,从活跃的gtp结合信号状态切换到不活跃的gtp结合信号状态。由于ras家族的三种亚型(hras、nras和kras)经常发生突变,而ras基因的突变会降低gtpase的活性或增加gaps的不敏感性,其反过来会激活ras蛋白,从而导致细胞生长失控诱发癌变。这些事实表明,ras在人类癌症的发生、发展过程中发挥了重要的作用,是治疗癌症的一个重要靶点。
3.hdac是一类催化赖氨酸去乙酰化的关键酶。研究显示它与基因转录、细胞增殖、血管生成、迁移、分化、转移等生物学过程密切相关,在直肠癌、胃癌、肝癌、乳腺癌和肺癌等多种肿瘤细胞中都有过表达现象。因此,hdac已成为抗肿瘤药物设计中的重要靶点。目前,已知的hdac有18种,根据其序列同源性可细分为四类:i类hdac,包含hdac1、2、3和8;ii类hdac,包含iia类的hdac4、5、7和9与iib类的hdac6和10;iii类hdac,包含sir2相关酶(sirtuins);iv类hdac,包含hdac11。通常,hdac抑制剂药效团可分为三部分:(1)帽基基团,用于识别hdac活性口袋;(2)锌离子结合基团,一般分为苯甲酰胺和羟肟酸两种,用于螯合hdac催化口袋底部的锌离子;(3)连接基团,用于连接锌离子结合基团与帽基基团。迄今为止,已有几种hdac抑制剂上市用于癌症治疗,如vorinostat(saha)、belinostat(pxd-101)、panobinostat(lbh589)和tacedinaline(ci994)等。不过、已上市的hdac抑制剂对实体瘤的治疗效果不甚理想,而且产生多种副作用,限制了它们在临床上的应用范围。因此,研发新型的hdac抑制剂作为抗肿瘤药物是十分有益的。
技术实现要素:
4.技术问题:本发明的目的是利用文献报道的以ras/raf为靶点的2,4,6-三甲氧基苯基-4-甲氧基-3-氨基苄基砜结构单元为帽基基团,以hdac为靶点的苯甲酰胺或羟肟酸结构单元为锌离子结合基团,通过线性基团连接,得到一类含有ras/raf蛋白干扰基团的hdac抑制剂;本发明还披露这类化合物的制备方法。
5.技术方案:本发明的一种含有ras/raf蛋白干扰基团的hdac抑制剂是基于已知的
以ras/raf为靶点的2,4,6-三甲氧基苯基-4-甲氧基-3-氨基苄基砜(9)为帽基基团,其结构如式1所示,
[0006][0007]
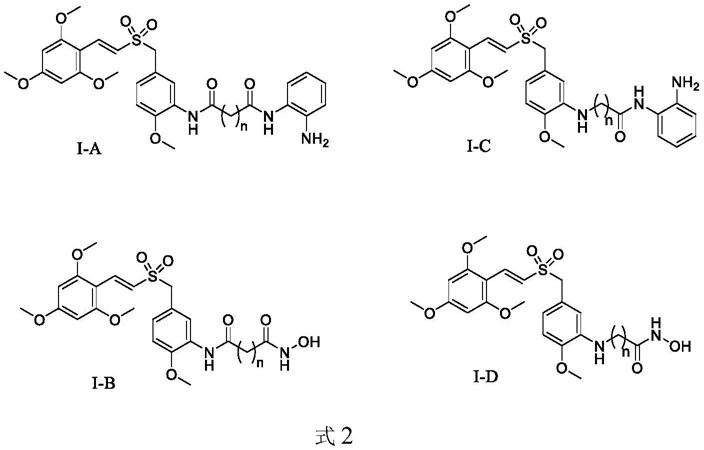
通过连接基团在其结构引入苯甲酰胺或羟肟酸结构单元,得到结构如式2所示的化合物即hdac抑制剂,分为以下具有代表性的i-a、i-b、i-c、i-d 4种化合物,
[0008][0009]
式2中,n=1-5。
[0010]
本发明的含有ras/raf蛋白干扰基团的hdac抑制剂的制备方法,其中,hdac抑制剂i-a按照以下反应式3-1进行制备:
[0011][0012]
其中,tbtu代表2-(1h-苯并三偶氮l-1-基)-1,1,3,3-四甲基脲四氟硼酸酯,et3n代表三乙胺,dmf代表n,n-二甲基甲酰胺。
[0013]
合成hdac抑制剂i-a具体采用如下步骤:
[0014]
将化合物13、14或15与1.0-1.5当量tbtu溶于无水dmf中,在室温下搅拌5-10min,加入1.0-1.5当量的et3n,再加入1.0-2.0当量的邻苯二胺,在氮气保护下,反应液于25-35℃下搅拌2-4h,然后减压除去溶剂,浓缩液经硅胶柱层析分离,洗脱液为dcm与ch3oh混合溶剂,分别对应得到化合物16、17或18淡黄色固体产物。
[0015]
hdac抑制剂i-b按照以下反应式3-2进行制备:
[0016][0017]
其中,tbtu代表2-(1h-苯并三偶氮l-1-基)-1,1,3,3-四甲基脲四氟硼酸酯,et3n代表三乙胺,dmf代表n,n-二甲基甲酰胺。
[0018]
合成hdac抑制剂i-b具体采用如下步骤:
[0019]
将化合物13、14或15与1.0-1.5当量tbtu溶于无水dmf中,在室温下搅拌5-10min,加入1.0-1.5当量的et3n,再加入1.0-2.0当量的盐酸羟胺,在氮气保护下,反应液于25-35℃下搅拌2-4h,然后减压除去溶剂,浓缩液经硅胶柱层析分离,洗脱液为dcm与ch3oh混合溶剂,分别对应得到化合物19、20或21淡黄色固体产物。
[0020]
hdac抑制剂i-c按照以下反应式3-3进行制备:
[0021][0022]
其中,tbtu代表2-(1h-苯并三偶氮l-1-基)-1,1,3,3-四甲基脲四氟硼酸酯,et3n代表三乙胺,dcm代表二氯甲烷。
[0023]
合成hdac抑制剂i-c具体采用如下步骤:
[0024]
将化合物28、29或30与1.0-1.5当量hatu溶于无水dcm中,在室温下搅拌5-10min,加入1.0-1.5当量的et3n,再加入1.0-2.0当量的邻苯二胺,在氮气保护下,反应液于25-35℃下搅拌2-4h,然后减压除去溶剂,浓缩液经硅胶柱层析分离,洗脱液为dcm与ch3oh混合溶剂,得到31、32或33淡黄色固体产物。
[0025]
hdac抑制剂i-d按照以下反应式进行制备:
[0026][0027]
其中,tbtu代表缩合剂2-(1h-苯并三偶氮l-1-基)-1,1,3,3-四甲基脲四氟硼酸酯,et3n代表三乙胺,dcm代表二氯甲烷。
[0028]
合成hdac抑制剂i-d具体采用如下步骤:
[0029]
将化合物28、29或30与1.0-1.5当量tbtu溶于无水dcm中,在室温下搅拌5-10min,加入1.0-1.5当量的et3n,再加入1.0-2.0当量的盐酸羟胺,在氮气保护下,反应液于25-35℃下搅拌2-4h,然后减压除去溶剂,浓缩液经硅胶柱层析分离,洗脱液为dcm与ch3oh混合溶剂,得到34、35或36淡黄色固体产物。
[0030]
进一步的,式1中化合物9及其衍生物13、14、15、28、29和30的制备按式4所示的反应式进行。
[0031][0032]
式4中,meoh代表甲醇,etoh代表乙醇,et3n代表三乙胺,dmf代表n,n-二甲基甲酰胺,dcm代表二氯甲烷,thf代表四氢呋喃;除特别指明外,n=1-5。
[0033]
化合物9及其衍生物13、14、15、28、29和30采用如下方法制备:
[0034]
1)化合物2-9根据文献方法(j.med.chem.2020,63,186
–
204)合成;
[0035]
2)化合物10、11或12的合成:将丁二酸单乙酯、戊二酸单甲酯或己二酸单甲酯溶解于dcm中,在0℃下加入草酰氯,室温搅拌过夜。减压蒸馏除去溶剂,再用dcm溶解后,加入化合物9,室温搅拌15min后逐滴滴加et3n,继续搅拌2h。反应完成后,用dcm萃取溶液,有机相用饱和食盐水洗涤三次,再用无水硫酸钠干燥后浓缩,得到黄色固体10、11或12。
[0036]
3)化合物13、14或15的合成:将化合物10、11或12溶解于etoh:h2o(2:1)溶液中,在室温下加入lioh
·
h2o,然后搅拌过夜。反应完成后,减压蒸馏除去溶剂,得到淡黄色固体,用少量水洗,过滤得到淡黄色固体13、14或15。
[0037]
4)化合物25、26或27的合成:将化合物9溶解于dmf中,室温下加入化合物溴乙酸乙
7、sgc7901、hepg2、a431的抗癌活性,具体结果见表2。目标化合物16-21和31-36均具有抗癌活性。值得注意的是化合物19和34对所测癌细胞表现出强的抗癌活性,其中化合物19的ic
50
值在0.084-1.68μm之间,化合物34的ic
50
值在0.045-0.99μm之间,明显高于母体化合物9的活性。与阳性对照saha(ic
50
值介于4.64至7.56μm之间)相比,化合物19和34的抗癌活性也明显更高。在所测试的六种癌细胞中,化合物19和34对非小细胞肺癌h1975细胞的细胞毒活性最强。构效关系表明,采用较长碳链连接基团所得化合物的抗癌活性比采用较短碳链连接基团所得化合物的抗癌活性要弱。此外,以羟肟酸作为锌离子结合基团的化合物19-21和34-36比以苯甲酰胺作为锌离子结合基团的化合物16-18和31-33具有更强的抗增殖活性。这可能是由于羟肟酸的结构分子具有较高的亲和力,更容易与锌离子结合,导致更高的细胞毒性。此外,还测定了这些化合物对人正常肝细胞lo2和脐静脉内皮细胞huvec的毒性。虽然化合物19和34对所测的任一癌细胞的毒性都高于阳性对照药物saha对癌细胞的毒性,但是它们对正常细胞的毒性低于saha。这表明所得的hdac抑制剂在提高抗肿瘤活性的同时,还降低了对正常细胞的毒性。
[0044]
我们还测试了化合物19和34对四种癌细胞panc-1、ht29、k562、mv4-11的抗癌活性,母体化合物9和hdac抑制剂saha为阳性对照,结果见表3。化合物34对所测癌的细胞表现出很强的抗癌活性,其ic
50
值在0.022-0.80μm之间,明显高于母体化合物9和药物saha的活性,也优于化合物19。
[0045]
根据上述结果,我们测试了化合物19和34对hdac的活性,结果见表4。化合物19和34对hdac 1、2、3、4、6和8均具有很好的抑制作用,其中对hdac1、2和3的ic
50
值在8-20nm区域内,而对hdac 4、6和8的ic
50
值则在60-90nm区域内。化合物19和34对hdac1、2和3的抑制作用明显高于hdac4和6,这表明它们可选择性地的抑制i类hdac。化合物19和34对hdac3表现出了显著的抑制效果,其ic
50
值分别为9.61nm和8.97nm。需要指出的是,saha对hdac8抑制活性达到微摩尔级别,几乎没有抑制作用,而化合物19和34对hdac8仍然具有中等强度的抑制作用,其ic
50
值分别为89.76nm和88.64nm。此外,化合物19和34对ii类hdac4和6的抑制效果也明显高于saha。综上,引入高活性帽基结构基团有助于提高对hdac的抑制作用。
[0046]
我们使用免疫共沉淀实验和免疫印迹分析,测试了化合物19和34对ras和raf蛋白表达的影响,结果见图1。在input组中,经过19和34处理的细胞,ras蛋白表达无明显变化,而p-raf(c-rafs
ser338
)蛋白的表达却明显下调。在ip组中,经过19和34处理的细胞,ras蛋白表达仅有轻微的下调,但p-raf(c-rafs
ser338
)蛋白的表达则明显下调,且下调效果优于阳性对照化合物9,而saha则对ras和raf蛋白的表达没有影响。结果表明,ras和raf是目标化合物19和34的靶向蛋白,它们可以阻断ras和raf蛋白之间的信号转导,从而导致p-raf蛋白表达下调。
[0047]
体外抗癌活性和hdac活性试验以及免疫共沉淀和免疫印迹实验结果表明,本发明所述的目标化合物对一些癌细胞具有显著的抑制作用,可用于制备药物。
附图说明
[0048]
图1.化合物对ras-raf蛋白的作用。
具体实施方式
[0049]
本发明以组蛋白去乙酰化酶(histone deacetylases,hdac)为靶点的羟肟酸或苯甲酰胺结构单元为锌离子结合基团,通过线性基团连接,得到一类含有ras/raf蛋白干扰基团的hdac抑制剂,以期获得高效低毒的抗肿瘤药物;本发明还披露了这类化合物的制备方法以及它们在制备抗肿瘤药物方面的应用。
[0050]
本发明由下述实施例进一步的说明,但这些说明并不限制本发明。除特别指出外,一些化合物如化合物2-9采用文献方法制备。按本发明方法制备的化合物经核磁氢谱和碳谱及高分辨质谱确定了化合物的分子结构。
[0051]
(一)、化合物9及其衍生物13、14、15、28、29和30的制备
[0052]
(1)化合物2的制备
[0053]
将化合物1(3.0g,18.0mmol)溶解于dmf(30.0ml)中,加入ch3i(3.8g,26.7mmol)和无水k2co3(6.2g,45.0mmol),室温搅拌过夜。通过tlc监测反应。反应完成后,减压蒸馏除去溶剂,得到粗产物。用dcm溶解粗产物,并用饱和食盐水溶液(100ml)洗涤三次。有机层用无水硫酸钠干燥后真空浓缩,得到白色固体。产率91.5%。
[0054]1h nmr(600mhz,cdcl3)δ9.95(s,1h),8.36(d,j=2.1hz,1h),8.10(dd,j=8.7,2.1hz,1h),7.25(d,j=8.7hz,1h),4.07(s,3h).
[0055]
(2)化合物3的制备
[0056]
将化合物2(1.01g,5.58mmol)溶解于20ml meoh中,在0℃下逐渐添加nabh4(0.27g,7.25mmol),并在相同温度下搅拌20分钟。使用tlc监测反应进程。反应完成后在0℃下滴加1m hcl溶液,减压蒸馏除去溶剂,获得白色固体粗产物。然后加入dcm(300ml)溶解粗产物,并用饱和食盐水溶液(100ml)洗涤三次。有机层用无水硫酸钠干燥后真空浓缩,得到白色固体。产率:100%。
[0057]1h nmr(600mhz,cdcl3)δ7.85(d,j=2.1hz,1h),7.54(dd,j=8.6,2.1hz,1h),7.08(d,j=8.6hz,1h),4.68(s,2h),3.96(s,3h),1.99(s,1h).
[0058]
(3)化合物4的制备
[0059]
将化合物3(600.0mg,3.28mmol)溶解于20ml dcm中,在0℃下逐渐添加pbr3(1.32g,4.92mmol),并在相同温度下搅拌3h,用tlc监测反应过程。反应完成后,在0℃下滴加1m nahco3溶液反应。将得到的固体产物用dcm溶解,用饱和食盐水溶液(100ml)洗涤三次,有机层用无水硫酸钠干燥后,真空浓缩,得到白色固体。产率99.3%。
[0060]1h nmr(600mhz,cdcl3)δ7.90(d,j=2.3hz,1h),7.58(dd,j=8.6,2.3hz,1h),7.07(d,j=8.7hz,1h),4.47(s,2h),3.97(s,3h).
[0061]
(4)化合物5的制备
[0062]
将化合物4(1g,4mmol)溶解于20ml meoh中,在0℃下添加tga(467.92mg,5mmol),和naoh(337.79mg,8.53mmol),反应混合物在室温下搅拌3h,通过tlc监测反应进程。反应完成后,在0℃下滴加1m hcl溶液,减压蒸馏除去溶剂,获得固体粗产物。用少量水洗,过滤得到淡黄色固体。产率90.1%。
[0063]1h nmr(600mhz,dmso)δ12.64(s,1h),7.83(d,j=2.2hz,1h),7.61(dd,j=8.6,2.3hz,1h),7.34(d,j=8.7hz,1h),3.92(s,3h),3.83(s,2h),3.14(s,2h).
[0064]
hr-ms(m/z)(esi):calcd for c
10h11
no2s,[m-h]-:256.04;found:256.0498.
[0065]
(5)化合物6的制备
[0066]
将化合物5(514.6mg,3.8mmol)溶解于20ml acoh中,在室温下逐滴添加30%的h2o2溶液(1.322g,38.8mmol),反应混合物在50℃下搅拌6h,通过tlc监测反应进程。反应完成后减压蒸馏除去溶剂,获得粗产物。以dcm:meoh(80:1-20:1)为洗脱剂,经硅胶层析法分离纯化,得淡黄色固体。产率71.4%。
[0067]1h nmr(600mhz,dmso)δ7.92(d,j=2.2hz,1h),7.68(dd,j=8.7,2.2hz,1h),7.43(d,j=8.8hz,1h),4.69(s,2h),4.22(s,2h),3.95(s,3h).
[0068]
hr-ms(m/z)(esi):calcd for c
10h11
no7s,[m-h]-:288.03;found:288.0324.
[0069]
(6)化合物8的制备
[0070]
将化合物6(514.6mg,3.8mmol)溶解于30ml甲苯中,依次加入哌啶(412μl,4.2mmol)、苯甲酸(650mg,5.2mmol)、化合物7(1.1g,5.7mmol),在40℃下搅拌回流过夜,通过tlc监测反应进程。反应完成后,溶液冷却至室温,过滤,得粗产物,用meoh重结晶,得黄色固体。产率36.3%。
[0071]1h nmr(600mhz,dmso)δ7.88(d,j=1.9hz,1h),7.64(dd,j=8.7,1.9hz,1h),7.50(d,j=15.6hz,1h),7.39(d,j=8.7hz,1h),7.09(d,j=15.6hz,1h),6.29(s,2h),4.55(s,2h),3.93(s,3h),3.85(s,9h).
[0072]
(7)化合物9的制备
[0073]
将化合物8(3.60g,8.6mmol)溶解于20ml etoh中,依次加入h2o(10ml),nh4cl(2.27g,5.2mmol),fe粉(2.86g,50.1mmol),反应混合物在70℃下搅拌6h,通过tlc监测反应进程。在反应完成后,过滤除去固体残渣,获得的滤液减压蒸馏除去溶剂,得粗产物,再用dcm重结晶,得黄色固体。产率91.1%。
[0074]1h nmr(600mhz,dmso)δ7.59(d,j=15.7hz,1h),7.09(d,j=15.7hz,1h),6.74(d,j=8.2hz,1h),6.63(d,j=2.1hz,1h),6.49(dd,j=8.2,2.1hz,1h),6.29(s,2h),4.77(s,2h),4.21(s,2h),3.85(s,6h),3.84(s,3h),3.74(s,3h).
[0075]
hr-ms(m/z)(esi):calcd for c
19h23
no6s,[m h]
:394.13;found:394.1315.
[0076]
(8)化合物10的制备
[0077]
将丁二酸单乙酯(919.8mg,6.3mmol)溶解于20ml dcm溶液中,在0℃下加入草酰氯(2ml),反应混合物在室温下搅拌过夜。减压蒸馏去除溶剂获得无色油状物,用dcm(5ml)溶解后,在0℃下加至含有化合物9(2.25g,5.7mmol)的dcm(15ml)溶液中,搅拌15min后,逐滴加入et3n(761.7mg,1048.5μl),再继续搅拌2h,通过tlc监测反应进程。在反应完成后,用dcm(300ml)萃取溶液,有机相用饱和食盐水溶液(100ml)洗涤三次,再用无水硫酸钠干燥后,真空浓缩,得到黄色固体。产率86.2%.
[0078]1h nmr(600mhz,cdcl3)δ8.35(s,1h),7.87(s,1h),7.84(d,j=15.6hz,1h),7.15(dd,j=8.3,1.8hz,1h),7.09(d,j=15.6hz,1h),6.87(d,j=8.4hz,1h),6.08(s,2h),4.20(s,2h),4.16
–
4.13(m,2h),3.88(s,3h),3.84(s,3h),3.82(s,6h),2.69(dd,j=7.8,4.4hz,4h),1.26(t,j=7.1hz,3h).
[0079]
hr-ms(m/z)(esi):calcd for c
25h31
no9s,[m h]
:522.17;found:522.1800.
[0080]
(9)化合物11的制备
[0081]
按照上述化合物10的合成方法,使用草酰氯(2ml)、dcm(20ml)、戊二酸单甲酯
(6.3mmol)、et3n(1048.5μl)和化合物9(5.7mmol)获得化合物11。黄色固体,产率88.3%.
[0082]1h nmr(600mhz,cdcl3)δ8.37(s,1h),7.84(d,j=15.6hz,1h),7.72(s,1h),7.15(dd,j=8.4,2.0hz,1h),7.09(s,1h),6.87(d,j=8.4hz,1h),6.08(s,2h),4.21(s,2h),3.88(s,3h),3.84(s,3h),3.83(s,6h),3.68(s,3h),2.43(dd,j=13.7,7.0hz,4h),2.02(p,j=7.2hz,2h).
[0083]
hr-ms(m/z)(esi):calcd for c
25h31
no9s,[m-h]-:520.17;found:520.1772
[0084]
(10)化合物12的制备
[0085]
按照上述化合物10的合成方法,使用草酰氯(2ml)、dcm(20ml)、己二酸单甲酯(6.3mmol)、et3n(1048.5μl)和化合物9(5.7mmol)获得化合物12。黄色固体,产率85.4%.
[0086]1h nmr(600mhz,cdcl3)δ8.37(s,1h),7.84(d,j=15.6hz,1h),7.70(s,1h),7.15(dd,j=8.4,1.8hz,1h),7.11(d,j=15.6hz,1h),6.87(d,j=8.4hz,1h),6.08(s,2h),4.21(s,2h),3.88(s,3h),3.84(s,3h),3.83(s,6h),3.67(s,3h),2.39
–
2.34(m,4h),1.71(dd,j=6.9,4.1hz,4h).
[0087]
hr-ms(m/z)(esi):calcd for c
26h33
no9s,[m h]
:536.20;found:536.2076.
[0088]
(11)化合物13的制备
[0089]
将化合物10(1773.1mg,3.4mmol)溶解于etoh:h2o(2:1,30ml)中,在室温下加入lioh
·
h2o(357mg,8.5mmol),然后室温搅拌过夜,通过tlc监测反应进程。反应完成后,减压蒸馏除去溶剂,获得固体粗产物。用少量水洗,过滤得到淡黄色固体。产率97.0%.
[0090]1h nmr(600mhz,dmso)δ12.14(s,1h),9.16(s,1h),8.03(s,1h),7.59(d,j=15.6hz,1h),7.11(d,j=15.6hz,1h),7.04
–
6.99(m,2h),6.29(s,2h),4.34(s,2h),2.63(t,j=6.6hz,2h),2.47(d,j=6.6hz,2h).
[0091]
hr-ms(m/z)(esi):calcd for c
23h27
no9s,[m-h]-:492.16;found:492.1654.
[0092]
(12)化合物14的制备
[0093]
按照上述化合物13的合成方法,使用化合物11代替化合物10反应,得到淡黄色固体,产率91.0%.
[0094]1h nmr(600mhz,dmso)δ12.09(s,1h),9.09(s,1h),7.97(s,1h),7.59(d,j=15.6hz,1h),7.12(d,j=15.6hz,1h),7.04(dd,j=8.5,1.3hz,1h),7.01(d,j=8.5hz,1h),6.29(s,2h),4.35(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.40(t,j=7.3hz,2h),2.26(t,j=7.4hz,2h),1.81
–
1.74(m,2h).
[0095]
hr-ms(m/z)(esi):calcd for c
24h29
no9s,[m-h]-:506.18;found:506.1856.
[0096]
(13)化合物15的制备
[0097]
按照上述化合物13的合成方法,使用化合物12代替化合物10反应,得到淡黄色固体,产率90.5%.
[0098]1h nmr(600mhz,dmso)δ12.02(s,1h),9.06(s,1h),7.98(s,1h),7.59(d,j=15.6hz,1h),7.12(d,j=15.7hz,1h),7.04(dd,j=8.5,1.2hz,1h),7.01(d,j=8.5hz,1h),6.29(s,2h),4.34(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.37(t,j=6.7hz,2h),2.23(t,j=7.0hz,2h),1.58
–
1.50(m,4h).
[0099]
hr-ms(m/z)(esi):calcd for c
25h31
no9s,[m h]
:522.18;found:522.1822.
[0100]
(14)化合物25的制备
[0101]
将化合物9(393.4mg,1mmol)溶解于dmf(30ml)中,室温下依次加入化合物22(664.0mg,4mmol),k2co3(552.0mg,4mmol)和ki(66.4mg,0.4mmol),在氮气的保护下,反应混合溶液于70℃下搅拌3h,通过tlc监测反应进程。在反应完成后,用dcm(300ml)萃取溶液,有机相用饱和食盐水溶液(100ml)洗涤三次,再用无水硫酸钠干燥后,减压蒸馏,得到的固体以pe:ea:dcm(10:1:1-60:1:1)为洗脱剂,经硅胶层析法分离纯化,得黄色固体。产率80.0%.
[0102]1h nmr(600mhz,cdcl3)δ7.80(d,j=15.7hz,1h),7.26(s,1h),7.04(d,j=15.7hz,1h),6.73(d,j=8.1hz,1h),6.71(dd,j=8.1,1.9hz,1h),6.50(d,j=1.8hz,1h),6.08(s,2h),4.22(q,j=7.1hz,2h),4.16(s,2h),3.86(s,2h),3.85(s,3h),3.84(s,3h),3.82(s,6h),1.28(t,j=7.1hz,3h).
[0103]
hr-ms(m/z)(esi):calcd for c
23h29
no8s,[m h]
:480.16;found:480.1698.
[0104]
(15)化合物26的制备
[0105]
按照上述化合物25的合成方法,用化合物23代替化合物22反应,得到化合物26。黄色固体,产率82.1%.
[0106]1h nmr(600mhz,cdcl3)δ7.84(d,j=15.7hz,1h),7.06(d,j=15.7hz,1h),6.72(d,j=8.1hz,1h),6.69(dd,j=8.1,1.8hz,1h),6.62(d,j=1.6hz,1h),6.09(s,2h),4.17(s,2h),4.16
–
4.12(m,2h),3.84(s,3h),3.83(s,3h),3.82(s,6h),3.41(t,j=6.5hz,2h),2.57(d,j=6.5hz,2h),1.26(d,j=7.1hz,3h).
[0107]
hr-ms(m/z)(esi):calcd for c
24h31
no8s,[m h]
:494.18;found:494.1865.
[0108]
(16)化合物27的制备
[0109]
按照上述化合物25的合成方法,用化合物24代替化合物22反应,得到化合物27。黄色固体,产率84.2%.
[0110]1h nmr(600mhz,cdcl3)δ7.83(d,j=15.7hz,1h),7.05(d,j=15.7hz,1h),6.70(d,j=8.1hz,1h),6.65(dd,j=8.1,2.0hz,1h),6.58(d,j=1.9hz,1h),6.08(s,2h),4.22(s,1h),4.16(s,2h),4.14
–
4.10(m,2h),3.84(s,3h),3.82(s,3h),3.81(s,6h),3.11(t,j=6.9hz,2h),2.36(t,j=7.3hz,2h),1.91(dd,j=14.3,7.1hz,2h),1.24(d,j=7.1hz,3h).
[0111]
hr-ms(m/z)(esi):calcd for c
27h37
no8s,[m h]
:536.23;found:536.2331
[0112]
(17)化合物28的制备
[0113]
将化合物25(719.1mg,1.5mmol)溶解于thf:h2o(2:1;30ml)混合溶剂中,于室温下加入lioh
·
h2o(126mg,3mmol),然后搅拌24h,通过tlc监测反应进程。在反应完成后,溶液减压蒸馏除去溶剂,获得淡黄色粗产物。粗产物用少量水洗后,过滤得到淡黄色固体,产率89.4%.
[0114]1h nmr(600mhz,dmso)δ7.55(d,j=15.7hz,1h),7.11(d,j=15.7hz,1h),6.80(d,j=8.2hz,1h),6.58(dd,j=8.1,1.9hz,1h),6.42(d,j=2.0hz,1h),6.29(s,2h),5.75(s,1h),4.26(s,2h),3.85(s,6h),3.84(s,3h),3.79(s,3h),3.72(s,2h).
[0115]
hr-ms(m/z)(esi):calcd for c
21h25
no8s,[2m h]
:903.28;found:903.2828
[0116]
(18)化合物29的制备
[0117]
按照上述化合物28的合成方法,使用化合物26代替化合物25反应,得到淡黄色固体,产率87.6%.
[0118]1h nmr(600mhz,dmso)δ7.55(d,j=15.7hz,1h),7.11(d,j=15.7hz,1h),6.80(d,j=8.2hz,1h),6.58(dd,j=8.1,1.9hz,1h),6.42(d,j=2.0hz,1h),6.29(s,2h),5.75(s,1h),4.26(s,2h),3.85(s,6h),3.84(s,3h),3.79(s,3h),3.72(s,2h).
[0119]
hr-ms(m/z)(esi):caled for c
22h27
no8s,[m h]
:466.16;found:466.1568
[0120]
(19)化合物30的制备
[0121]
按照上述化合物28的合成方法,使用化合物27代替化合物25反应,得到淡黄色固体,产率88.4%。
[0122]
1h nmr(600mhz,dmso)δ12.01(s,1h),7.53(d,j=15.7hz,1h),7.11(d,j=15.7hz,1h),6.75(d,j=8.2hz,1h),6.52(dd,j=8.1,1.9hz,1h),6.47(d,j=1.9hz,1h),6.29(s,2h),4.73(s,1h),4.28(s,2h),3.84(s,9h),3.76(s,3h),2.91(t,j=6.8hz,2h),2.17(t,j=7.4hz,2h),1.50
–
1.42(m,4h),1.24(dd,j=10.3,4.9hz,2h).
[0123]
hr-ms(m/z)(esi):calcd for c
25h33
no8s,[m h]
:508.20;found:508.2035.
[0124]
(二)、目标化合物的制备
[0125]
实施例1.化合物16的制备
[0126]
将化合物13(250.0mg,0.51mmol)溶解于dmf(30ml)中,室温下依次加入tbtu(245.6mg,0.76mmol)、et3n(106.4μl,0.765mmol)和邻苯二胺(108.0mg,1.01mmol),在氮气保护下,反应混合物在35℃搅拌2h。在反应完成后,用dcm(300ml)萃取溶液,有机相用饱和食盐水溶液(100ml)洗涤三次,再用无水硫酸钠干燥,减压蒸馏,所得固体以dcm:meoh(60:1-30:1)为洗脱剂,经硅胶层析法分离纯化,得淡黄色固体,产率36.2%.
[0127]1h nmr(600mhz,dmso)δ9.20(s,1h),9.19(s,1h),8.02(s,1h),7.60(d,j=15.6hz,1h),7.15(dd,j=7.7,0.9hz,1h),7.12(d,j=15.6hz,1h),7.06
–
7.03(m,1h),7.01(d,j=8.4hz,1h),6.89(td,j=8.0,1.4hz,1h),6.71(dd,j=8.0,1.2hz,1h),6.53(td,j=7.7,1.2hz,1h),6.29(s,2h),4.98(s,2h),4.35(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.73(t,j=6.9hz,2h),2.63(t,j=7.0hz,2h).
13
c nmr(151mhz,dmso)δ171.12,170.91,164.11,161.43(c
×
2),149.88,142.49,133.38,127.72,127.45,126.31,126.00,124.78,124.01,123.84,121.10,116.56,116.15,111.26,103.05,91.40(c
×
2),60.34,56.53(c
×
2),56.22,56.09,32.01,31.36.
[0128]
hr-ms(m/z)(esi):calcd for c
29h33
n3o8s,[m h]
:584.20;found:584.2095.
[0129]
实施例2.化合物17的制备
[0130]
按照上述化合物16的合成方法,使用化合物14代替化合物13反应,得到淡黄色固体,产率38.4%.
[0131]1h nmr(600mhz,dmso)δ9.13(s,1h),9.12(s,1h),8.03(s,1h),7.60(d,j=15.6hz,1h),7.19-7.17(m,1h),7.13(d,j=15.6hz,1h),7.05(dd,j=8.4,1.2hz,1h),7.01(d,j=8.5hz,1h),6.91
–
6.88(m,1h),6.72(dd,j=8.0,1.2hz,1h),6.55(td,j=7.8,1.2hz,1h),6.29(s,2h),4.91(s,2h),4.35(s,2h),3.86(s,6h),3.84(s,3h),3.82(s,3h),2.45(t,j=7.2hz,2h),2.38(t,j=7.4hz,2h),1.88(dd,j=14.6,7.3hz,2h).
13
c nmr(151mhz,dmso)δ171.45,171.26,164.12,161.43(c
×
2),150.00,142.32,133.41,127.61,127.70,126.20,125.87,124.99,124.03,124.02,121.08,116.65,116.35,111.26,103.05,91.41(c
×
2),60.33,56.53(c
×
2),56.22,56.08,35.91,35.52,21.84.
[0132]
hr-ms(m/z)(esi):calcd for c
30h35
n3o8s,[m h]
:598.20;found:598.2320.
[0133]
实施例3.化合物18的制备
[0134]
按照上述化合物16的合成方法,使用化合物15代替化合物13反应,得到淡黄色固体,产率37.6%.
[0135]1h nmr(600mhz,dmso)δ9.14(s,1h),9.07(s,1h),8.01(s,1h),7.60(d,j=15.6hz,1h),7.16(dd,j=7.8,0.9hz,1h),7.13(d,j=15.6hz,1h),7.05(d,j=8.2hz,1h),7.01(d,j=8.5hz,1h),6.91
–
6.88(m,1h),6.73(dd,j=7.9,1.0hz,1h),6.57
–
6.53(m,1h),6.29(s,2h),4.92(s,2h),4.35(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.42(s,2h),2.34(d,j=6.0hz,2h),1.63(s,4h).
13
c nmr(151mhz,dmso)δ171.67,171.52,164.11,161.42(c
×
2),150.02,142.21,133.39,127.69,127.52,126.19,125.78,125.02,124.10,124.02,121.09,116.77,116.44,111.25,103.05,91.40(c
×
2),60.31,56.52(c
×
2),56.21,56.08,36.36,36.12,25.52.25.43
[0136]
hr-ms(m/z)(esi):calcd for c
31h37
n3o8s,[m h]
:612.20;found:612.2515.
[0137]
实施例4.化合物19的制备
[0138]
将化合物13(208.0mg,0.42mmol)溶解于dmf(30ml)中,室温下依次加入tbtu(175.3mg,0.55mmol)、et3n(350.6μl,2.52mmol)和nh2oh
·
hcl(116.7mg,1.68mmol),在氮气保护下,反应混合物在35℃搅拌2h,通过tlc监测反应进程。在反应完成后,用dcm(300ml)萃取溶液,有机相用饱和食盐水溶液(100ml)洗涤三次,再用无水硫酸钠干燥,减压蒸馏,得到的固体以dcm:meoh(50:1-20:1)为洗脱剂,经硅胶层析法进一步分离纯化,得黄色固体。产率60.9%.
[0139]1h nmr(600mhz,dmso)δ10.43(s,1h),9.17(s,1h),8.74(s,1h),8.03(s,1h),7.59(d,j=15.6hz,1h),7.11(d,j=15.6hz,1h),7.01(t,j=7.5hz,2h),6.29(s,2h),4.34(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.63(t,j=7.1hz,2h),2.26(t,j=7.1hz,2h).
13
c nmr(151mhz,dmso)δ170.77,168.91,164.11,161.42(c
×
2),149.77,133.38,127.73,127.40,124.62,123.99,121.07,111.22,103.03,91.40(c
×
2),60.35,56.53(c
×
2),56.22,56.08,31.82,28.01.
[0140]
hr-ms(m/z)(esi):calcd for c
23h28
n2o9s,[m h]
:509.16;found:509.1625.
[0141]
实施例5.化合物20的制备
[0142]
按照上述化合物19的合成方法,使用化合物14代替化合物13反应,得到黄色固体,产率62.3%。
[0143]1h nmr(600mhz,dmso)δ10.40(s,1h),9.08(s,1h),8.71(d,j=1.1hz,1h),8.00(s,1h),7.59(d,j=15.6hz,1h),7.12(d,j=15.6hz,1h),7.04(dd,j=8.5,1.1hz,1h),7.01(d,j=8.5hz,1h),6.29(s,2h),4.35(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.37(t,j=7.3hz,2h),2.00(t,j=7.4hz,2h),1.79
–
1.76(m,2h).
13
c nmr(151mhz,dmso)δ171.30,169.24,164.12,161.43(c
×
2),150.00,133.39,127.90
–
127.69,127.60,124.98,124.03,121.07,111.26,103.04,91.41(c
×
2),60.30,56.53(c
×
2),56.23,56.09,35.86,32.15,21.83.
[0144]
hr-ms(m/z)(esi):calcd for c
24h30
n2o9s,[m h]
:523.16;found:523.1737
[0145]
实施例6.化合物21的制备
[0146]
按照上述化合物19的合成方法,使用化合物15代替化合物13反应,得到黄色固体,产率63.6%.
[0147]1h nmr(600mhz,dmso)δ10.38(s,1h),9.05(s,1h),8.70(s,1h),7.99(s,1h),7.59(d,j=15.6hz,1h),7.12(d,j=15.6hz,1h),7.02(dd,j=19.4,8.3hz,2h),6.29(s,2h),4.34(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h),2.37(s,2h),1.97(s,2h),1.52(s,4h).
13
c nmr(151mhz,dmso)δ171.63,169.46,164.12,161.42(c
×
2),150.01,133.39,127.60,125.00,124.01,121.08,111.25,103.04,91.40(c
×
2),60.30,56.53(c
×
2),56.16,36.26,32.64,25.35.
[0148]
hr-ms(m/z)(esi):calcd for c
25h32
n2o9s,[m h]
:537.16;found:537.1925.
[0149]
实施例7.化合物31的制备
[0150]
将化合物28(225.6mg,0.5mmol)溶解于dmf(30ml)中,室温下依次加入hatu(247.1mg,0.65mmol)、et3n(90.3μl,0.65mmol)和邻苯二胺(162.15mg,1.5mmol),在氮气保护下,反应混合溶液于35℃下搅拌2h,通过tlc监测反应进程。在反应完成后,用dcm(300ml)萃取溶液,有机相用饱和食盐水溶液(100ml)洗涤三次,再用无水硫酸钠干燥,减压蒸馏,所得固体以dcm:meoh(100:1-80:1)为洗脱剂,经硅胶层析法进一步分离纯化,得淡黄色固体。产率37.0%.
[0151]1h nmr(600mhz,dmso)δ9.18(s,1h),7.60(d,j=15.7hz,1h),7.18(dd,j=7.8,1.2hz,1h),7.11(d,j=15.7hz,1h),6.92
–
6.89(m,1h),6.83(d,j=8.2hz,1h),6.72(dd,j=8.0,1.2hz,1h),6.61(dd,j=8.1,1.8hz,1h),6.54(dd,j=7.5,1.2hz,1h),6.52(d,j=1.9hz,1h),6.29(s,2h),5.33(s,1h),4.87(s,2h),4.29(s,2h),3.86(s,2h),3.85(s,6h),3.84(s,3h),3.82(s,3h).
13
c nmr(151mhz,dmso)δ169.00,164.08,161.40(c
×
2),147.10,142.66,137.68,133.22,126.56,126.18,124.15,119.90,116.61,116.18,112.72,109.95,91.44(c
×
2),60.93,56.52(c
×
2),56.08,55.95,47.03.
[0152]
hr-ms(m/z)(esi):calcd for c
27h31
n3o7s,[m h]
:542.20;found:542.2022
[0153]
实施例8.化合物32的制备
[0154]
按照上述化合物31的合成方法,使用化合物29代替化合物28反应,得到淡黄色固体,产率38.4%.
[0155]1h nmr(600mhz,dmso)δ9.19(s,1h),7.59(d,j=15.7hz,1h),7.14(dd,j=15.8,8.2hz,2h),6.92
–
6.89(m,1h),6.78(d,j=8.2hz,1h),6.72(d,j=7.9hz,1h),6.61(d,j=1.5hz,1h),6.58
–
6.53(m,2h),6.29(s,2h),4.94(d,j=40.8hz,3h),4.30(s,2h),3.85(s,6h),3.83(s,3h),3.77(s,3h),3.29(t,j=6.4hz,2h),2.62(t,j=6.7hz,2h).
13
c nmr(151mhz,dmso)δ170.46,164.05,161.38(c
×
2),147.02,142.46,138.00,133.14,126.39,126.00,124.12,123.66,121.90,119.27,116.60,116.26,112.62,109.82,103.04,91.38(c
×
2),60.90,56.50(c
×
2),56.06,55.85,39.97,35.67.
[0156]
hr-ms(m/z)(esi):calcd for c
28h33
n3o7s,[m h]
:556.22;found:556.2161.
[0157]
实施例9.化合物33的制备
[0158]
按照上述化合物31的合成方法,使用化合物30代替化合物28反应,得到淡黄色固体,产率39.3%.
[0159]1h nmr(600mhz,dmso)δ9.08(s,1h),7.55(d,j=15.7hz,1h),7.16(dd,j=7.8,
1.1hz,1h),7.12(d,j=15.7hz,1h),6.89(t,j=7.6hz,1h),6.75(d,j=8.1hz,1h),6.72(d,j=9.1hz,1h),6.55
–
6.52(m,2h),6.50(d,j=1.6hz,1h),6.29(s,2h),4.79(d,j=76.4hz,3h),4.28(s,2h),3.84(s,6h),3.84(s,3h),3.76(s,3h),2.95(t,j=7.0hz,2h),2.30(t,j=7.4hz,2h),1.58
–
1.53(m,4h),1.34
–
1.30(m,2h).
13
c nmr(151mhz,dmso)δ171.57,164.05,161.39(c
×
2),146.79,142.29,138.24,133.19,126.15,125.72,124.14,124.09,121.97,118.76,116.69,116.39,112.23,109.74,103.10,91.40(c
×
2),60.97,56.49(c
×
2),56.07,55.80,43.08,36.26,28.80,26.83,25.59.
[0160]
hr-ms(m/z)(esi):calcd for c
31h39
n3o7s,[m h]
:598.26;found:598.2642.
[0161]
实施例10.化合物34的制备
[0162]
将化合物28(189.6mg,0.42mmol)溶解于dcm(30ml)中,室温下依次加入tbtu(175.3mg,0.55mmol)、et3n(350.6μl,2.52mmol)和nh2oh
·
hcl(116.7mg,1.68mmol),在氮气保护下,反应混合物在35℃搅拌2h,通过tlc监测反应进程。在反应完成后,用dcm(300ml)萃取溶液,有机相用饱和食盐水溶液(100ml)洗涤三次,再用无水硫酸钠干燥,减压蒸馏,所得固体以dcm:meoh(60:1-20:1)为洗脱剂,经硅胶层析法进一步分离纯化,得黄色固体。产率52.6%。
[0163]1h nmr(600mhz,dmso)δ10.63(s,1h),8.90(s,1h),7.60(s,1h),7.10(d,j=15.6hz,1h),6.81(d,j=8.2hz,1h),6.60(d,j=8.0hz,1h),6.46(d,j=23.1hz,1h),6.29(s,2h),5.09(s,1h),4.27(s,2h),3.85(s,6h),3.85(s,3h),3.80(s,3h),3.55(s,2h).
13
c nmr(151mhz,dmso)δ166.93,164.08,161.41(c
×
2),147.10,137.63,133.20,124.10,121.75,119.93,112.76,109.89,103.05,91.43(c
×
2),60.90,56.51(c
×
2),56.08,55.92,44.40.
[0164]
hr-ms(m/z)(esi):calcd for c
21h26
n2o8s,[m h]
:467.14;found:467.1579.
[0165]
实施例11.化合物35的制备
[0166]
按照上述化合物31的合成方法,使用化合物29代替化合物28反应,得到淡黄色固体,产率51.2%。
[0167]1h nmr(600mhz,dmso)δ10.49(s,1h),8.82(s,1h),7.58(d,j=15.7hz,1h),7.13(d,j=15.7hz,1h),6.78(d,j=8.0hz,1h),6.56(d,j=9.2hz,2h),6.30(s,2h),4.92(s,1h),4.30(s,2h),3.86(d,j=3.0hz,9h),3.77(s,3h),3.19(t,j=6.6hz,2h),2.26(t,j=6.8hz,2h).
13
c nmr(151mhz,dmso)δ168.29,164.04,161.37(c
×
2),146.92,137.81,133.10,124.08,121.86,119.25,112.47,109.77,103.00,91.37(c
×
2),60.78,56.50(c
×
2),56.07,55.81,39.87,32.30.
[0168]
hr-ms(m/z)(esi):calcd for c
22h28
n2o8s,[m h]
:481.16;found:481.1663.
[0169]
实施例12.化合物36的制备
[0170]
按照上述化合物31的合成方法,使用化合物30代替化合物28反应,得到淡黄色固体,产率50.2%。
[0171]1h nmr(600mhz,dmso)δ10.36(s,1h),8.70(s,1h),7.55(d,j=15.7hz,1h),7.12(d,j=15.7hz,1h),6.75(d,j=8.2hz,1h),6.52(dd,j=8.1,1.7hz,1h),6.48(d,j=1.7hz,1h),6.29(s,2h),4.72(s,1h),4.28(s,2h),3.85(s,9h),3.76(s,3h),2.92(dd,j=12.7,6.6hz,2h),1.93(t,j=7.4hz,2h),1.47(ddt,j=22.9,15.2,7.4hz,4h),1.24
–
1.20
(m,2h).
13
c nmr(151mhz,dmso)δ169.53,164.05,161.38(c
×
2),146.75,138.20,133.16,124.09,121.94,118.74,112.17,109.69,103.04,91.36(c
×
2),60.90,56.48(c
×
2),56.08,55.77,43.05,32.76,28.63,26.76,25.42.
[0172]
hr-ms(m/z)(esi):calcd for c
25h34
n2o8s,[m h]
:523.21;found:523.2129.
[0173]
(三)、化合物体外细胞毒活性测试
[0174]
实验方法:取对数生长期的细胞计数,接种于96孔培养板内,每孔约8000-10000个细胞。培养过夜,待细胞贴壁后进行给药,分别设给药组和对照组。待测的目标化合物用dmso溶液配制成贮液,临用前用细胞培养基稀释成一系列浓度,其中dmso的终浓度不超过4
‰
(下面实验类同)。每个浓度设3个复孔。加药后培养72h,加20μl浓度为5mg/ml的mtt,37℃孵育4h,弃去上清,加入150μl的dmso溶解。用酶标仪在490纳米波长下测定每孔的od值,并计算抑制率,做浓度-抑制率曲线计算ic
50
值。
[0175]
实验例1.
[0176]
测试了目标化合物对非小细胞肺癌细胞a549和h1975、胃癌细胞sgc-7901、肝癌细胞hepg2、乳腺癌细胞mcf-7和表皮癌细胞a431的抗增殖活性以及它们对人正常肝细胞lo2和脐静脉内皮细胞huvec的毒性,母体化合物9和hdac抑制剂saha作为阳性对照。观察化合物在不同浓度下对肿瘤细胞生长的抑制情况,计算抑制率及其ic
50
值来评价药物的细胞毒活性,结果见表2。
[0177]
表2.化合物的体外抗癌活性及对正常细胞的毒性
[0178][0179][0180]
实验例2.
[0181]
测试了目标化合物19和34对胰腺癌细胞panc-1、结肠癌细胞ht29、慢性粒细胞白血病癌细胞k562和人髓性单核细胞白血病细胞mv4-11的抗增殖活性,母体化合物9和hdac抑制剂saha作为阳性对照。观察化合物在不同浓度下对肿瘤细胞生长的抑制情况,计算抑制率及其ic
50
值来评价药物的细胞毒活性,结果见表3。
[0182]
表3.化合物19和34对一些癌细胞的ic
50
值
[0183][0184]
(四)、化合物对hdac的抑制活性检测
[0185]
实验方法:根据人组蛋白脱乙酰基酶hdac试剂盒说明书(上海抚生实业有限公司生产),应用双抗体夹心法测定样品中人组蛋白脱乙酰基酶(hdac1、2、3、4、6、8)水平。分别设空白孔(空白对照孔不加样品及酶标试剂,其余各步操作相同)、标准孔、待测样品孔。将不同浓度的药物与hdac亚型蛋白混合加于酶标板孔底部,用封板膜封板后置37℃温育30min。用洗涤液洗涤5次后,于每孔中加入酶标试剂50μl,空白孔除外。再封板后置37℃温育30min,洗涤5次。然后每孔先加入显色剂a 50μl,再加入显色剂b 50μl,轻轻震荡混匀,37℃避光显色10min。最后每孔加终止液50μl,终止反应。以空白孔调零,用酶标仪在450nm波长依序测量各孔的吸光度(od值)。并计算抑制率,做浓度-抑制率曲线计算ic
50
值。
[0186]
实验例3.
[0187]
测试了目标化合物19和34对六种hdac亚型的抑制情况,即hdac1、hdac2、hdac3、hdac4、hdac6、hdac8。以hdac抑制剂saha为阳性对照,观察化合物在不同浓度下对hdac酶的抑制情况,计算抑制率及其ic
50
值来评价化合物的酶活性,结果见表4。
[0188]
表4.化合物对人组蛋白去乙酰化酶hdac的抑制活性
[0189][0190]
(五)、化合物对ras和raf蛋白作用的检测
[0191]
实验方法:采用免疫共沉淀和免疫印迹检测的实验方法测试化合物对ras和raf蛋白的作用。取对数生长期的h1975细胞计数,接种于6孔培养板内,每孔约100000-1000000个细胞,培养过夜,待细胞贴壁后进行给药处理24h。消化细胞,转移至离心管中,加入预冷的细胞裂解液,置于4℃摇床平台上,温和振荡15min;然后在10,000rpm,4℃的条件下离心5min,取上清液转移至新的离心管中。此时将上清液分为两组,一组为ip组,在上清液中加入ras抗体,4℃慢摇过夜。加入充分重悬的g蛋白琼脂糖,4℃慢摇4h。2500rpm离心5min,小心吸除上清,pbs洗涤沉淀5次。完成洗涤后,去除上清,加入1xsds-page电泳上样缓冲液重悬,混匀后,95℃水浴10min,作为样品蛋白,然后通过免疫印迹的方法来检测。另一组作为input组,直接在上清液中加入1xsds-page电泳上样缓冲液重悬,混匀后,95℃水浴10min,作为样品蛋白,然后再通过免疫印迹的实验方法来检测。
[0192]
首先配制分离胶和浓缩胶,待胶成型后,吸取适量样品蛋白加入样品孔中,在样品旁的孔中加入预染的蛋白marker,未添加样品上清的孔中,加入1x sds上样缓冲液保持胶面平衡。打开电源,,电压开始设置为60v,开始电泳,当蛋白样品进入分离胶后,电压可提高到120v。电泳完成后,开始转pvdf膜,设置电流流1.1a,电压25v,时间30min进行半干转。转膜结束后,取出pvdf膜并作好标记,用tbst溶液洗涤3次,每次10min。接着将pvdf膜放入平皿中,加入封闭液,摇床振荡1.5-2h。封闭结束后,用tbst溶液洗涤。接着将膜放入含一抗的孵育盒中,4℃摇床振荡孵育过夜。第二天取出,室温振荡30min,吸弃一抗,tbst洗涤3次。二抗孵育,室温摇床振荡反应2h。二抗反应结束后用tbst洗涤。将ecl化学发光试剂盒中的a、b两种液体按l:l等体积混合,配置成工作液备用。将pvdf膜从tbst中取出,滴加适量工作液,使用g:box chemixr5显色成像
[0193]
实验例4.
[0194]
测试了化合物19和34对ras和raf蛋白的作用,以化合物9和saha作为阳性对照。观察不同化合物在同等浓度下对ras和raf蛋白表达的影响,结果见图1。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。