1.本发明属于制药工程技术领域,具体涉及一种双氯芬酸钠的合成方法。
背景技术:
2.双氯芬酸钠,又称双氯灭痛,是一种衍生于芳基乙酸类的非甾体类消炎镇痛药。由瑞士汽巴-嘉基公司研制,于1974年上市。临床上用于解热、镇痛,并广泛用于各类风湿性关节炎、红斑狼疮、神经炎及癌症、手术后引起的疼痛和各种原因引起的发热等。双氯灭痛主要是通过抑制体内前列腺素的合成发挥作用,与其他消炎镇痛药相比,其抗炎作用强大,比消炎痛(吲哚美辛)强2~2.5倍,比阿司匹林强20~50倍;口服吸收迅速,排泄快,长期服用无积蓄作用;个体差异小,在抗风湿性药物中处于领先地位,并获得了广泛使用。其结构式如(i)所示:
[0003][0004]
美国专利3558690、英国专利1132128以及费声钱等(医药工业,1979,10,14)叙述了以邻(氯)苯甲酸为起始原料经ullmann缩合和脱羧反应制得关键中间体2,6二氯苯胺,继而经过酰化、关环、水解开环、成盐得到双氯芬酸钠(i)。该方法原料易得,是我国最早用于生产双氯芬酸钠的合成路线,但是步骤冗长,合成过程用到大毒、及强腐蚀性化合物,环境污染严重,且总收率低。
[0005]
荷兰专利6604752和日本专利23418叙述了以溴苯和2,6二氯苯胺进行ullmann缩合反应,一步制得2,6二氯二苯胺,收率接近50%。此方法一度为很多工厂所采用,但因路线中有少量难以分离的n—苯基-2-氯-6-溴苯胺等副产物生成,这些微量的芳香溴化物的存在会引起胃溃疡等副作用,因此该路线的工业应用受到很大限制。
[0006]
德国专利1815802叙述了以草酰氯为酰化试剂,通过stoll合成法得到中间体n-芳基吲哚-2,3-二酮,再经过还原、水解得到双氯芬酸钠。该路线虽然收率高,但使用了价格昂贵,高毒性、强腐蚀性及刺激性的草酰氯,三废污染严重,劳动保护要求高,且步骤冗长。
[0007]
日本专利描述了以邻氯苯甲酸和2,6-二氯苯胺为原料进行缩合,再使用重氮甲烷或氰化钠等为碳源得到双氯芬酸钠。该路线使用了易爆的重氮甲烷或剧毒品氰化钠,操作危险,环境污染严重,不适合工业生产。
[0008]
欧洲专利0380712,世界专利022522描述了以苯胺为原料,经酰化、醚化、chapman重排制备双氯芬酸钠的方法。陈芬儿等对此方法进行了重大改进。陈芬儿(中国医药工业杂志,1998,29,339)、美国专利4978773、秦丙昌(应用化工,2008,3,275)描述了以2,6-二氯二苯胺和氯乙酰氯经酰化、分子内傅克烷基化、水解开环得目标产物。该方法是目前国内生产
双氯芬酸钠的主要路线之一,但反应步骤较多,且原子经济性差,三废污染也较为严重。
[0009]
陈芬儿等(中国专利cn1580039a)叙述了以环己酮为原料,经氯化、羧化、氢化还原、缩合、芳构化、成盐等得到双氯芬酸钠。该路线虽然收率较高,但使用到氯气、氯乙酰氯、有机磷等毒性大、腐蚀性和刺激性强的物料,三废污染严重,劳动保护要求高,且步骤冗长。
[0010]
中国专利cn108947861a叙述了以苯乙酸为起始原料,经硝化、还原、酰胺化、缩合、重排、水解等步骤制得目标产物。该路线步骤较长,且使用到腐蚀性和刺激性的硝化试剂,以及较为昂贵的钯催化剂,不适合工业化生产。
[0011]
以上这些现有的双氯芬酸钠的合成路线,要么需要使用毒性大刺激性大的原料,三废污染严重,劳动保护要求高,要么步骤繁琐、收率低,或是中间体价格昂贵不适合工业化生产。由此可见,寻找一条收率高、原子经济性高、绿色环保、成本低及操作简便的新的合成路线,对于满足当前双氯芬酸钠的生产需求以及环境保护具有重大意义。
技术实现要素:
[0012]
本发明的目的在于克服现有技术的不足,而提供一种原子经济性高、步骤少、总收率高、成本低和环境友好的双氯芬酸钠的合成方法。
[0013]
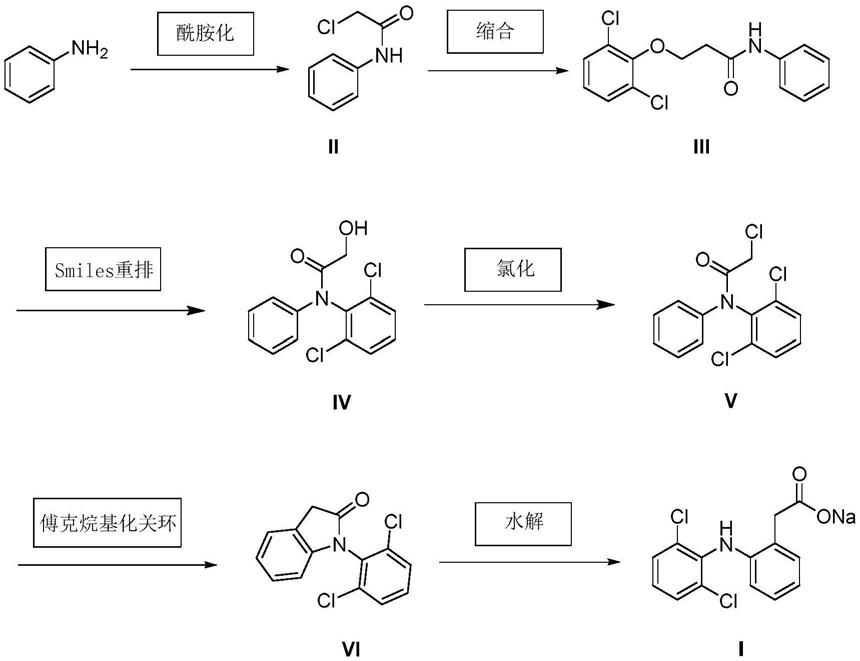
本发明提供的双氯芬酸钠的合成方法,其合成路线如下:
[0014][0015]
合成的具体步骤为:
[0016]
(1)苯胺和氯乙酸在硼酸类催化剂的作用下于有机溶剂中进行酰胺化反应,得到化合物2-氯-n-苯基乙酰胺(ii);
[0017]
(2)化合物2-氯-n-苯基乙酰胺(ii)与2,6-二氯苯酚在碳酸钾和相转移催化剂作用下发生缩合反应,得到化合物2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii);
[0018]
(3)在碱性催化剂作用下,化合物2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)经smiles重排反应,得到化合物n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv);
[0019]
(4)化合物n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)与二氯亚砜在催化剂作用下发生氯化反应,得到化合物n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v);
[0020]
(5)化合物n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)在路易斯酸催化剂作用下发生傅克烷基化关环反应,得到化合物1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi);
[0021]
(6)化合物1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)经无机碱水解反应,即得目标产物双氯芬酸钠(i)。
[0022]
由苯胺计算,总收率可达74%。产品质量符合usp、bp、日本药局和中国药典各项标准。
[0023]
优选的,在步骤(1)中,所述酰胺化反应的催化剂为含氟取代的芳基硼酸、邻卤代芳基硼酸、氨基硼酸、硼酸和硼酸酯中的任一种。
[0024]
优选的,在步骤(1)中,控制苯胺与氯乙酸、催化剂的摩尔比为1:(1~5):(0.001~0.5)。
[0025]
优选的,在步骤(1)中,所述酰胺化反应所使用的有机溶剂为氯苯、甲苯、二甲苯、二氯苯、均三甲苯、乙腈和正丁醇中的任一种。
[0026]
优选的,在步骤(1)中,控制所述酰胺化反应温度为70~170℃,反应时间为1~20h。
[0027]
优选的,在步骤(2)中,所用相转移催化剂为聚乙二醇400、聚乙二醇600、苄基三乙基氯化铵和四丁基溴化铵中的任一种。
[0028]
优选的,在步骤(2)中,控制2-氯-n-苯基乙酰胺(ii)与2,6-二氯苯酚、碳酸钾、相转移催化剂的投料摩尔比为1:(0.5~2):(1~5):(0.001~0.5)。
[0029]
优选的,在步骤(2)中,所述缩合反应所使用的有机溶剂为氯苯、甲苯、二甲苯、二氯苯、均三甲苯、乙腈和正丁醇中的任一种。
[0030]
优选的,在步骤(2)中,控制所述缩合反应温度为80~160℃,反应时间为2~15h。
[0031]
优选的,在步骤(3)中,所述smiles重排反应的催化剂为无机碱或有机碱,其中,无机碱为氢氧化钠、氢氧化钙、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾和钠氢中的任一种;有机碱为三乙胺、吡啶、甲醇钠、二异丙基胺基锂和1,8-二氮杂双环[5.4.0]十一碳-7-烯中的任一种。
[0032]
优选的,在步骤(3)中,控制2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)与催化剂的摩尔比为1:(0.001~20)。
[0033]
优选的,在步骤(3)中,所述smiles重排反应所使用的有机溶剂为n,n-二甲基甲酰胺、二甲基亚砜、甲醇、乙醇、甲苯、乙腈、乙酸乙酯和丙酮中的任一种。
[0034]
优选的,在步骤(3)中,控制所述smiles重排反应温度为0~100℃,反应时间为0.5~10h。
[0035]
优选的,在步骤(4)中,所述催化剂为吡啶、n,n-二甲基苯胺、三乙胺和n,n-二甲基甲酰胺中的任一种。
[0036]
优选的,在步骤(4)中,控制n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)与二氯亚砜、催化剂的摩尔比为1:(1~4):(0.001~0.5)。
[0037]
优选的,在步骤(4)中,所述氯化反应所使用的有机溶剂为n,n-二甲基甲酰胺、二甲基亚砜、氯化亚砜、甲苯、乙腈、二氯甲烷、1,2-二氯乙烷、乙酸乙酯和丙酮中的任一种。
[0038]
优选的,在步骤(4)中,控制所述氯化反应温度为10~100℃,反应时间为0.5~10h。
[0039]
优选的,在步骤(5)中,所述路易斯酸催化剂为三氯化铝、三氯化铁、氯化锌、溴化锌和氯化锡中的任一种。
[0040]
优选的,在步骤(5)中,控制n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)与路易斯酸催化剂的摩尔比为1:(0.5~5)。
[0041]
优选的,在步骤(5)中,所述傅克烷基化关环反应所使用的有机溶剂为二氯甲烷、1,2-二氯乙烷、氯苯、甲苯、二甲苯、二氯苯、正丁醇和二苯醚中的任一种。
[0042]
优选的,在步骤(5)中,所述傅克烷基化关环反应可不使用溶剂。
[0043]
优选的,在步骤(5)中,控制所述傅克烷基化关环反应温度为50~200℃,反应时间为1~10h。
[0044]
优选的,在步骤(6)中,所述无机碱为碳酸钠、碳酸氢钠和氢氧化钠中的任一种。
[0045]
优选的,在步骤(6)中,所述水解反应可不需要相转移催化剂。
[0046]
优选的,在步骤(6)中,使用相转移催化剂可以促进所述水解反应的进行;所述相转移催化剂为苄基三乙基氯化铵或四丁基溴化铵。
[0047]
优选的,在步骤(6)中,控制1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)与无机碱、相转移催化剂的摩尔比为1:(0.1~10):(0~0.5)。
[0048]
优选的,在步骤(6)中,所述水解反应所用有机溶剂为甲苯、二甲苯、二苯醚、甲醇和乙醇中的任一种。
[0049]
优选的,在步骤(6)中,所述水解反应可为无溶剂反应。
[0050]
优选的,在步骤(6)中,控制所述水解反应温度为10~130℃,反应时间为1~10h。
[0051]
有益效果:
[0052]
本发明所使用的酰化试剂为氯乙酸,相较于现有路线中使用的氯乙酰氯,氯乙酸的毒性低、刺激性和腐蚀性小、劳动保护要求低;使用硼酸类化合物催化酰胺化反应,催化效果好,反应转化率高,可循环利用,且硼酸类催化剂也较为绿色环保。
[0053]
在现有的路线中,2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)经过smile重排后会发生水解变为2,6-二氯二苯胺,这就使得后续需要再次使用氯乙酰氯进行酰胺化,造成原子经济性差,污染严重以及劳动保护要求高等问题。本发明与现有技术相比,通过将smile重排的产物控制在中间体n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv),而不发生水解副反应,这样就避免再次使用氯乙酰氯,大大提高原子经济,而且本发明方法不再需要使用强刺激性腐蚀性原料氯乙酰氯,劳动保护要求低,环境友好以及原子经济性高。
[0054]
本发明设计的合成方法,原料价廉易得,使用各种无机碱和高效催化剂,总收率高(达74%),环境友好,原子经济性高,成本低,劳动保护要求低,产品质量符合usp、bp、日本药局和中国药典各项标准,有效解决了现有方法的弊端,适合于工业化生产。
附图说明
[0055]
图1为n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺的1h-nmr。
[0056]
图2为n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺的高分辨质谱。
具体实施方式
[0057]
为更好的说明本发明的目的、技术方案和优点和更加清楚明白,下面将结合具体实施例对本发明作进一步说明,但并不因此限制本发明的内容。
[0058]
(一)2-氯-n-苯基乙酰胺(ii)的制备
[0059]
实施例1
[0060]
将氯乙酸(10.395g,0.11mol)以及硼酸(61.83mg,0.001mol)、甲苯(200ml)置于干燥反应瓶中,缓慢加入苯胺(9.313g,0.1mol)。加料完毕后升至130℃,回流搅拌6h,tlc监测反应。反应完毕后加入碳酸氢钠中和反应液至中性,萃取分液,有机相用硫酸钠溶液(10%)洗涤,浓缩,减压蒸馏得黄色固体为2-氯-n-苯基乙酰胺(ii)(16.28g,96%),m.p.85~87℃。
[0061]
实施例2
[0062]
将氯乙酸(14.17g,0.15mol)以及硼酸(61.83mg,0.001mol)、甲苯(200ml)置于干燥反应瓶中,缓慢加入苯胺(9.313g,0.1mol)。加料完毕后升至130℃回流搅拌6h,tlc监测反应。反应完毕后加入碳酸氢钠中和反应液至中性,萃取分液,有机相用硫酸钠溶液(10%)洗涤,浓缩,减压蒸馏得黄色固体为2-氯-n-苯基乙酰胺(ii)(16.62g,98%),m.p.85~87℃。
[0063]
实施例3
[0064]
将氯乙酸(51.97g,0.55mol)以及硼酸(618.3mg,0.01mol)、甲苯(200ml)置于干燥反应瓶中,缓慢加入苯胺(46.565g,0.5mol)。加料完毕后升至140℃回流搅拌12h,tlc监测反应。反应完毕后加入碳酸氢钠中和反应液至中性,有机相用硫酸钠溶液(10%)洗涤,浓缩,减压蒸馏得黄色固体为2-氯-n-苯基乙酰胺(ii)(81.41g,96%),m.p.85~87℃。
[0065]
(二)2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)的制备
[0066]
实施例4
[0067]
将2-氯-n-苯基乙酰胺(ii)(16.69g,0.1mol)、2,6-二氯苯酚(16.63,0.1mol)、碳酸钾(27.64,0.2mol)、peg-400(0.4g)和二甲苯(160ml)置于反应瓶中,升温至150℃,回流搅拌2h,tlc监测反应。反应毕,用稀盐酸调至ph中性,萃取分液,有机相用无水硫酸钠干燥,减压蒸馏得黄色固体为2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)(29.46g,95%),m.p.78~80℃。
[0068]
实施例5
[0069]
将2-氯-n-苯基乙酰胺(ii)(33.38g,0.2mol)、2,6-二氯苯酚(33.26,0.2mol)、碳酸钾(69.1,0.5mol)、peg-600(1.8g)和二甲苯(320ml)置于反应瓶中,升温至150℃,回流搅拌4h,tlc监测反应。反应毕,用稀盐酸调至ph中性,萃取分液,有机相用无水硫酸钠干燥,减压蒸馏得黄色固体为2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)(59.55g,96%),m.p.78~80℃。
[0070]
(三)n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)的制备
[0071]
实施例6
[0072]
将2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)(31.02g,0.1mol)、无水碳酸钠
(10.60g,0.1mol)置于反应瓶中,氮气保护下,加入甲苯(100ml),升温至60℃进行反应,tlc监测反应。反应毕,用氯化铵调至ph中性,萃取分液,有机相用无水硫酸钠干燥,减压蒸馏得淡黄色固体为n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)(26.06g,85%),m.p.112~114℃。
[0073]
实施例7
[0074]
将2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)(31.02g,0.1mol)、无水碳酸钾(13.82g,0.1mol)置于反应瓶中,氮气保护下,加入甲苯(100ml),升温至70℃进行反应,tlc监测反应。反应毕,用氯化铵调至ph中性,萃取分液,有机相用无水硫酸钠干燥,减压蒸馏得淡黄色固体为n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)(26.65g,88%),m.p.112~114℃。
[0075]
实施例8
[0076]
将2-(2,6二氯苯氧基)-n-苯基乙酰胺(iii)(31.02g,0.1mol)、无水碳酸钠(5.3g,0.05mol)置于反应瓶中,氮气保护下,加入甲苯(100ml),升温至70℃进行反应,tlc监测反应。反应毕,用氯化铵调至ph中性,萃取分液,有机相用无水硫酸钠干燥,减压蒸馏得淡黄色固体为n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)(26.65g,88%),m.p.112~114℃。
[0077]
(四)n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)的制备
[0078]
实施例9
[0079]
将n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)(59.23g,0.2mol)、二氯亚砜(47.59,0.4mol)、二氯甲烷(100ml)置于干燥反应瓶中,边搅拌边缓慢滴加三乙胺(0.202g,0.002mol),回流反应4h,tlc监测反应。反应毕,过滤除去不溶物,减压回收二氯亚砜。得淡黄色固体为n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)(61.97g,99%),m.p.142~143℃。
[0080]
实施例10
[0081]
将n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)(59.23g,0.2mol)、二氯亚砜(100ml)置于干燥反应瓶中,边搅拌边缓慢滴加三乙胺(0.202g,0.002mol),回流反应3h,tlc监测反应。反应毕,过滤除去不溶物,减压回收二氯亚砜。得淡黄色固体为n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)(59.47g,95%),m.p.142~143℃。
[0082]
实施例11
[0083]
将n-(2,6-二氯苯基)-2-羟基-n-苯基乙酰胺(iv)(29.62g,0.1mol)、二氯亚砜(23.79,0.2mol)、二氯乙烷(100ml)置于干燥反应瓶中,边搅拌边缓慢滴加三乙胺(1.01g,0.01mol),升温至50℃反应1h,tlc监测反应。反应毕,过滤除去不溶物,减压回收二氯亚砜。得淡黄色固体为n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)(30.67g,98%),m.p.142~143℃。
[0084]
(五)1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)的制备
[0085]
实施例12
[0086]
将n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)(31.3g,0.1mol)置于干燥反应瓶中,氮气保护下加入三氯化铝(26.67g,0.2mol)升温至160℃,熔融状态反应6h,tlc监测反应。反应毕,趁热将反应液倒入冰水中得大量固体。过滤,滤液用二氯甲烷反萃浓缩后与固体合并,用去离子水洗涤,无水硫酸钠干燥,得棕黄色固体为1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)(26.69g,96%),m.p.124~125℃。
[0087]
实施例13
[0088]
将n-(2,6-二氯苯基)-2-氯-n-苯基乙酰胺(v)(31.3g,0.1mol)置于干燥反应瓶中,加入氯苯(50ml),搅拌,氮气保护下加入三氯化铝(26.67g,0.2mol),升温至160℃,反应4h,tlc监测反应。反应毕,向反应液中缓慢加入稀盐酸,萃取分液,有机相经硫酸钠固体干燥,过滤,减压浓缩后得棕黄色固体为1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)(19.46g,97%),m.p.124~125℃。
[0089]
(六)双氯芬酸钠(i)的制备
[0090]
实施例14
[0091]
将氢氧化钠(16g,0.4mol)和水(38ml)置于反应瓶中,搅拌溶解,冷却后,于搅拌下加1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)(27.7g,0.1mol)和二甲苯(100ml)、tebac(2.28g,0.01mol),升温回流6h。反应毕,冷却至室温,减压回收溶剂,加入水100ml重结晶,活性炭脱色后得白色固体为目标化合物双氯芬酸钠(i)(28.53g,95%),m.p.283~284℃。
[0092]
实施例15
[0093]
将氢氧化钠(16g,0.4mol)和水(38ml)置于反应瓶中,搅拌溶解,冷却后,于搅拌下加入1-(2,6-二氯苯基)-1,3-二氢-2h-吲哚-2-酮(vi)(27.7g,0.1mol)和甲醇(100ml),升温回流6h。反应毕,冷却至室温,减压回收甲醇,加入水100ml重结晶,活性炭脱色后得白色固体为目标化合物双氯芬酸钠(i)(30.12g,96%),m.p.283~284℃。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。