一种碘促进氧化法制备4-碘代n-芳基吡唑类化合物的方法
技术领域
1.本发明涉及有机合成技术领域,特别是涉及一种碘促进氧化法制备4-碘代n-芳基吡唑类化合物的方法。
背景技术:
2.目前,超过半数的化合物及药物属于杂环化合物。吡唑是一类重要的五元含氮杂环化合物,广泛存在于天然产物和生物活性分子中,是新药研发中构筑生物活性化合物的重要砌块,同时也是生物碱天然产物全合成中的重要的骨架单元。n-芳基吡唑作为其中重要的组成部分,一直以来都是研究的热点,在此结构基础上进行衍生,得到的新化合物将可能体现不同的生物活性,有利于农药或者医药先导的发现。例如,农药氟虫腈,由拜耳公司发现,具有杀虫谱广,能防治蔬菜、水稻和棉花等鳞翅目害虫,有杀螨活性,许多国内外公司以氟虫腈结构为先导,创制并成功上市了多个高效杀虫活性的衍生物;药物塞来昔布(celebrex),是用于缓解骨关节炎的症状和体征、缓解成人类风湿关节炎的症状和体征、治疗成人急性疼痛。
3.目前已经开发出许多用于合成n-芳基吡唑类化合物的方法,其中最常见的合成策略是:通过n-芳基肼与1,3-二羰基化合物或其类似物的环缩合反应得到。因而发展一种原料廉价易得、步骤简单、操作方便,条件温和、效率高的吡唑类化合物一直以来,是有机化学及药学工作者所关注的热点。因此,通过n-芳基-3-吡唑烷酮的脱氢芳构化/碘代的方法,来合成吡唑衍生物的方法仍然是具有相当的意义与价值。
技术实现要素:
4.本发明的目的是提供一种反应条件温和,操作步骤和后处理过程简单,产率高,适合于大量制备4-碘代n-芳基吡唑类化合物的方法。
5.为实现上述目的,本发明提供了如下方案:
6.本发明提供一种碘促进氧化法制备4-碘代n-芳基吡唑类化合物的方法,包括以下步骤:以n-芳基-3-吡唑烷酮类化合物为起始原料,以二甲基亚砜(dmso)为反应溶剂,碘为催化剂和碘代试剂,于100-120℃加热的反应条件下,n-芳基-3-吡唑烷酮类化合物发生脱氢芳构化/碘代串联,制得4-碘代n-芳基吡唑类化合物。
7.进一步地,所述n-芳基-3-吡唑烷酮类化合物的结构式见式1,式1中r1=h、2-me、2-cl、2,4-cl、3-cl、3-br、3-ome、4-me、4-f、4-cl、4-br或4-cf3;r2=h或ph;
[0008][0009]
进一步地,所述4-碘代n-芳基吡唑类化合物的结构式见式2,式2中r1=h、2-me、2-cl、2,4-cl、3-cl、3-br、3-ome、4-me、4-f、4-cl、4-br或4-cf3;r2=h或ph;
[0010][0011]
进一步地,反应时间为18-24h。
[0012]
进一步地,n-芳基-3-吡唑烷酮类化合物与碘的质量比为(32.4-48.0):55.8。
[0013]
进一步地,反应在空气氛围下进行。
[0014]
进一步地,于100-120℃反应结束后,还包括萃取、干燥和柱层析分离的过程。
[0015]
进一步地,反应结束后,冷却至室温,然后加入饱和硫代硫酸钠水溶液、乙酸乙酯(体积比1:1),萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物。
[0016]
本发明以n-芳基-3-吡唑烷酮类化合物为初始原料(0.20mmol,32.4mg-48.0mg),然后加入二甲基亚砜溶剂溶解,碘为催化剂和碘代试剂,于100-120℃温度下,在空气的反应条件下,反应18-24小时(薄层层析色谱监测),冷却至室温,然后加入水,乙酸乙酯(体积比1:1)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯),得到n-芳基吡唑类化合物(收率28%-88%)。本发明具有原料廉价易得,反应条件温和,官能团兼容性较好,操作步骤和后处理过程简单,效率较高;反应无需任何过渡金属催化剂等复杂反应条件的特点,避免了药用生物活性分子引入有害的金属杂质对药品的污染;同时通过芳构化/碘代反应在吡唑环上引入了碘原子,产物能够进一步应用于金属催化的偶联反应以及卤素/金属交换反应,易于衍生化,合成具有结构多样化的n-芳基吡唑类化合物,为相关分子的药物化学及生物活性研究,提供了研究基础。
[0017]
本发明制备得到的4-碘代n-芳基吡唑类化合物可以应用于药物中间体的合成。
[0018]
本发明公开了以下技术效果:
[0019]
1.该反应条件温和、无需过渡金属催化剂,反应在100-120℃条件下进行,操作简单,有利于大量制备4-碘代n-芳基吡唑类化合物。
[0020]
2.该反应具有较好的原子经济性,产物收率高。
[0021]
3.该反应底物范围广,官能团兼容性强。
附图说明
[0022]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0023]
图1为本发明制备方法的合成路线图;
[0024]
图2为实施例1中4-碘代n-芳基吡唑类化合物2a的合成路线图;
[0025]
图3为实施例2中4-碘代n-芳基吡唑类化合物2b的合成路线图;
[0026]
图4为实施例3中4-碘代n-芳基吡唑类化合物2c的合成路线图;
[0027]
图5为实施例4中4-碘代n-芳基吡唑类化合物2d的合成路线图;
7.41(m,2h),7.22-7.18(m,1h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.8,139.4,132.6,129.6,125.3,116.8,49.2.
[0047]
实施例2
[0048]
4-碘代n-芳基吡唑类化合物2b的合成(合成路线图见图3):
[0049]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1b(0.20mmol,35.4mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应18小时(用薄层层析色谱监测)。待原料1b反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2b(52.0mg,收率87%)。1h nmr(400mhz,dmso-d6):δ10.87(s,1h),8.37(s,1h),7.56-7.53(m,2h),7.24-7.22(m,2h),2.29(s,3h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.5,137.2,134.5,132.3,129.9,116.8,48.6,20.4.
[0050]
实施例3
[0051]
4-碘代n-芳基吡唑类化合物2c的合成(合成路线图见图4):
[0052]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1c(0.20mmol,36.0mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应20小时(薄层层析色谱监测)。待原料1c反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2c(40.4mg,收率66%)。1h nmr(400mhz,dmso-d6):δ10.93(s,1h),8.42(s,1h),7.72-7.68(m,2h),7.32-7.27(m,2h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.8,159.6(d,j
c-f
=240.2hz),136.0(d,j
c-f
=2.6hz),132.7,118.7(d,j
c-f
=8.2hz),116.2(d,j
c-f
=22.9hz),49.2;
19
f nmr(376mhz,dmso-d6):δ-117.85to-117.92(m,1f).
[0053]
实施例4
[0054]
4-碘代n-芳基吡唑类化合物2d的合成(合成路线图见图5):
[0055]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1d(0.20mmol,39.2mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应20小时(薄层层析色谱监测)。待原料1d反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2d(54.2mg,收率85%)。1h nmr(400mhz,dmso-d6):δ11.03(s,1h),8.48(s,1h),7.71-7.67(m,2h),7.51-7.47(m,2h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.9,138.2,132.8,129.4,129.1,118.3,50.0.
[0056]
实施例5
[0057]
4-碘代n-芳基吡唑类化合物2e的合成(合成路线图见图6):
[0058]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡
7.51(m,1h),7.48-7.44(m,1h),7.44-7.39(m,1h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.6,137.5,136.8,130.6,129.2,128.3,127.5,127.3,48.1.
[0068]
实施例9
[0069]
4-碘代n-芳基吡唑类化合物2i的合成(合成路线图见图10):
[0070]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1i(0.20mmol,39.2mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应22小时(薄层层析色谱监测)。待原料1i反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2i(53.5mg,收率83%)。1h nmr(400mhz,dmso-d6):δ11.08(s,1h),8.53(s,1h),7.77-7.76(m,1h),7.66-7.64(m,1h),7.47-7.43(m,1h),7.26-7.23(m,1h);
13
c{1h}nmr(100mhz,dmso-d6):δ163.0,140.4,134.0,133.1,131.2,124.8,116.4,115.1,50.5.
[0071]
实施例10
[0072]
4-碘代n-芳基吡唑类化合物2j的合成(合成路线图见图11):
[0073]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1j(0.20mmol,48.0mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应18小时(薄层层析色谱监测)。待原料1j反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2j(52.0mg,收率71%)。1h nmr(400mhz,dmso-d6):δ11.09(br,s,1h),8.53(s,1h),7.90(s,1h),7.71-7.66(m,1h),7.41-7.37(m,2h);
13
c{1h}nmr(100mhz,dmso-d6):δ163.0,140.5,133.1,131.5,127.7,122.4,119.2,115.5,50.5.
[0074]
实施例11
[0075]
4-碘代n-芳基吡唑类化合物2k的合成(合成路线图见图12):
[0076]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1k(0.20mmol,46.0mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应18小时(薄层层析色谱监测)。待原料1k反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2k(44.7mg,收率63%)。化合物2k可以用于减肥药利莫那班(rimonabant)其类似物的合成。1h nmr(400mhz,dmso-d6):δ10.94(s,1h),8.06(s,1h),7.84(s,1h),7.55(d,j=1.0hz,2h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.8,136.9,136.6,132.5,130.1,128.6,128.4,128.2,48.4.
[0077]
实施例12
[0078]
4-碘代n-芳基吡唑类化合物2l的合成(合成路线图见图13):
[0079]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1l(0.20mmol,38.4mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上
反应管盖,将其转移至100℃油浴锅中,在空气的反应条件下,反应24小时(薄层层析色谱监测)。待原料1l反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2l(20.0mg,收率32%)。化合物12产率较低的原因:部分原料在反应过程中发生分解。1h nmr(400mhz,dmso-d6):δ10.96(br,s,1h),8.48(s,1h),7.35-7.31(m,1h),7.26-7.24(m,2h),6.78-6.76(m,1h),3.79(s,3h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.6,160.2,140.5,132.8,130.4,111.1,108.8,102.3,55.4,49.3.
[0080]
实施例13
[0081]
4-碘代n-芳基吡唑类化合物2m的合成(合成路线图见图14):
[0082]
在25ml的反应管中,加入磁力搅拌子和碘(1.1equiv,55.8mg),称取n-芳基-3-吡唑烷酮类化合物1m(0.20mmol,47.6mg)加入,然后加入二甲基亚砜溶剂(2.0ml)溶解,盖上反应管盖,将其转移至120℃油浴锅中,在空气的反应条件下,反应24小时(用薄层层析色谱监测)。待原料1m反应完全后,反应管冷却至室温,然后加入饱和硫代硫酸钠溶液(5.0ml),乙酸乙酯(5.0ml)萃取3次,取有机层溶液,无水硫酸钠干燥,蒸干溶剂,然后将粗产物经柱层析分离(洗脱剂,石油醚:乙酸乙酯=5:1,体积比),得到n-芳基吡唑类化合物2m(20.1mg,收率28%)。化合物13产率较低的原因:部分原料在反应过程中发生分解。1h nmr(400mhz,dmso-d6):δ10.89(s,1h),7.41-7.39(m,3h),7.31-7.18(m,5h),7.11-7.09(m,2h);
13
c{1h}nmr(100mhz,dmso-d6):δ162.1,143.7,139.7,130.1,129.9,129.0,128.8,128.6,126.7,124.2,52.7.
[0083]
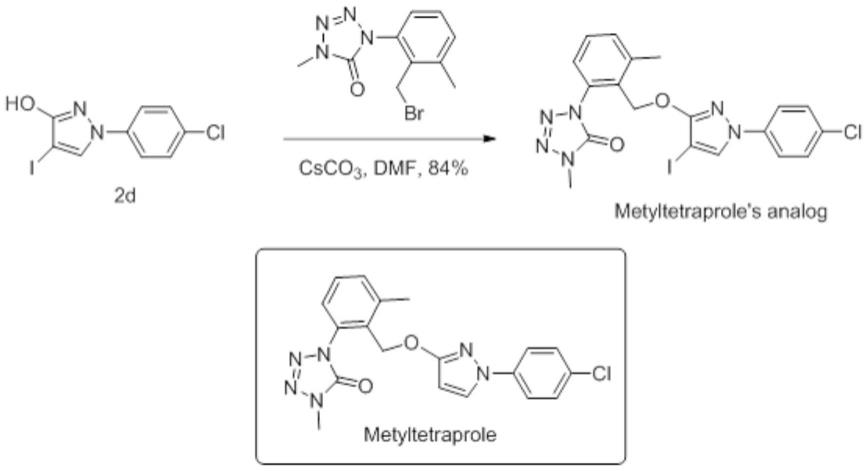
本发明制备得到的4-碘代n-芳基吡唑类化合物可以应用于药物中间体的合成,例如化合物2d可用于吡唑类杀菌剂metyltetraprole类似物的合成,路线图如图15所示。
[0084]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
