1.本发明属于药物合成领域,具体涉及一种富马酸伏诺拉生的制备方法。
背景技术:
2.胃酸相关疾病(ards)是一类胃酸与发病机理密切相关的上消化道疾病,包括胃食管反流病、消化不良、胃肠道溃疡、胃炎、十二指肠炎、及一些抗炎类药引发的消化道疾病。ards的发病率在全球范围内呈逐年上升趋势。在中国,ards已经成为重大疾病之一,严重影响了患者的生活质量,并给患者带来了较大的经济负担。在临床上同质子泵抑制剂(ppis)已被广泛应用于ards的治疗,是人类疾病的主要治疗药物。
3.富马酸伏诺拉生作为钾离子竞争性阻滞剂,体外活性实验表明该化合物抑制质子泵的能力是兰索拉唑的400倍,其相对于na
,k
-atpase的选择性在500倍以上,在体内有更强的效能和更持久的抑酸作用。
4.富马酸伏诺拉生作为新型口服抗胃酸药,武田制药已于2015年上市。其化学名为:1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基]-n-甲基甲胺富马酸盐,结构如下:
[0005][0006]
现有报道的富马酸伏诺拉生的制备主要有以下合成路线:
[0007]
武田原研专利cn101300229公开的富马酸伏诺拉生制备路线如下:
[0008][0009]
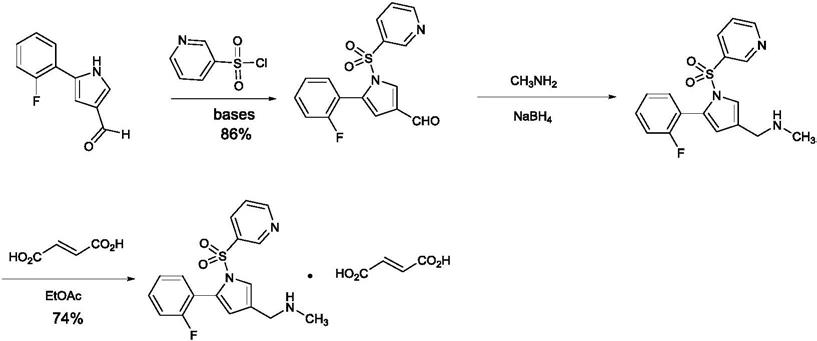
以5-(2-氟苯基)-1h-吡咯-3-甲醛为原料,以乙腈为溶剂,n,n-二异丙基乙胺为缚酸剂、4-二甲胺基吡啶为催化剂,与3-吡啶磺酰氯反应生成中间体5-(2-氟苯基)-1-(吡啶-3-磺酰基)-1h-吡咯-3-甲醛,再与甲胺反应生成席夫碱,硼氢化钠还原得到伏诺拉生游离碱,再与富马酸成盐,但是该反应路线中副反应多,杂质难以去除,需要多次精制且收率只
有49.9%,无法得到高纯度的富马酸伏诺拉生。
[0010]
该产品还原胺化生产过程中,会生成如下几个杂质极难去除:
[0011][0012]
cn104211618报道了以5-(2-氟苯基)-1h-吡咯-3-腈为原料的合成路线:
[0013][0014]
该路线中氰基转化到醛基需要用到雷尼镍催化加氢,工艺生产存在很大的安全隐患,同时产生的杂质i、ii、iii难以去除。
[0015]
文献j.med.chem.2012,55(9),4446-4456和专利cn105085484报道了以苯基吡咯和甲胺醇反应生成亚胺,经硼氢化钠还原,再进行boc酸酐保护得到中间体((5-苯基-1-h-吡咯-3-基)-n-甲基)甲基氨基甲酸叔丁酯,再与3-吡啶磺酰氯反应,最后脱boc保护得到伏诺拉生游离碱,再与富马酸成盐得到富马酸伏诺拉生:
[0016][0017]
该路线通过增加boc保护氨基,去除了反应过程中的杂质iii,纯度有所提高,但杂质i和杂质ii无法去除,且磺酰化反应中用到了钠氢高危试剂,安全性大大减弱,同样不利于工业化大生产。
技术实现要素:
[0018]
为弥补富马酸伏诺拉生现有合成工艺的技术缺陷,本发明公开了一种新的合成工艺路线,以市场易得、价格便宜的化合物为原料,反应条件温和,选择性高,所用试剂少,后
处理简单,可以稳定的得到高纯度,高收率产品,稳定的供应富马酸伏诺拉生。
[0019]
本发明合成路线如下:
[0020][0021]
具体步骤:
[0022]
(1)化合物1和化合物2在有机溶剂中,在催化剂和碱试剂的作用下发生酰化反应,生成化合物3;
[0023]
(2)化合物3与甲胺经还原胺化反应得到化合物4;
[0024]
(3)化合物4与二碳酸二叔丁酯反应,经酸洗后得到化合物5;
[0025]
(4)化合物5在酸性条件下脱boc得到化合物4;
[0026]
(5)化合物4与富马酸成盐得到化合物6即富马酸伏诺拉生。
[0027]
在一些实施方案中,步骤(1)中催化剂为dmap,与化合物1的摩尔比为(0.1~1)∶1,优选0.2∶1;化合物2与化合物1的摩尔比为(1~2)∶1,优选1.1∶1;采用的溶剂为极性非质子溶剂,优选四氢呋喃、乙腈、甲苯、二氯甲烷;采用的碱试剂选自三乙胺、dipea、碳酸钠,优选三乙胺,碱试剂与化合物1的摩尔比为(1.1~1.8)∶1。
[0028]
在一些实施方案中,步骤(2)中甲胺与化合物3的摩尔比为(1~4)∶1,优选(2~3)∶1;采用的溶剂为极性质子或非质子溶剂,优选甲醇、乙醇、四氢呋喃、乙腈;采用的还原剂选自硼氢化钠、醋酸硼氢化钠、氰基硼氢化钠,优选硼氢化钠,还原剂与化合物3的摩尔比为(0.5~1)∶1。
[0029]
在一些实施方案中,步骤(3)中二碳酸二叔丁酯与化合物4的摩尔比为(0.9~2.0)∶1,优选(1.0~1.5)∶1;溶剂为极性质子或非质子溶剂,优选二氯甲烷、甲苯;反应温度为15℃-45℃,优选20℃-30℃。后处理酸选用弱酸,选自低浓度的柠檬酸水溶液、稀盐酸,氢溴酸,乙酸,优选ph为2-3的稀盐酸溶液。
[0030]
在一些实施方案中,步骤(4)采用的酸选自浓盐酸、三氟乙酸、氯化氢甲醇溶液、氯化氢乙醇溶液、氯化氢异丙醇溶液、氯化氢乙酸乙酯溶液,优选浓盐酸,与化合物5的摩尔比为(2.0~6.0)∶1,优选(4.0~5.0)∶1。
[0031]
在一些实施方案中,步骤(5)中富马酸与化合物4的摩尔比为(1.0~2.0)∶1,优选(1.0~1.2)∶1,更优选(1.05~1.15)∶1;将富马酸先溶于极性质子溶剂后再加料,溶剂优选甲醇或乙醇。
[0032]
在一些实施方案中还包含精制步骤,精制体系选用甲醇水的体积混合比例为1.0∶(0.5~2.0),优选1.0∶(0.8~1.2),析晶温度为-10℃~20℃,优选温度为0℃~10℃。
[0033]
有益效果:
[0034]
本发明合成方法的起始物料简单易得,条件温和,不使用钠氢等高危试剂,后处理简单,收率与现有工艺基本持平,但有效去除了反应过程中产生的各种顽固性杂质(杂质i、杂质ii、杂质iii),提高了富马酸伏诺拉生的纯度(目前文献报道的hplc最高纯度为99.7%,本发明得到产品hplc纯度稳定在99.9%以上),品质高于目前市场上的原料药质量,保证了原料药的质量,间接减少了下游制剂产品的副作用,提高了制剂的有效性和安全性。
附图说明
[0035]
图1为实施例1化合物6富马酸伏诺拉生的hplc谱图;
[0036]
图2为实施例1化合物6富马酸伏诺拉生的核磁共振氢谱图;
[0037]
图3为原研参比制剂富马酸伏诺拉生的hplc谱图
具体实施方式
[0038]
以下对本发明进一步阐释,但并不限制本发明。
[0039]
实施例1:
[0040]
化合物3的制备
[0041][0042]
向500ml反应瓶中依次加入530.00g乙腈、200.00g的化合物1、25.83g的dmap、117.67g的三乙胺,开启搅拌,控温在15~35℃之间,滴加206.52g的化合物2,约30min左右滴加完毕;在15~35℃之间,保温反应1~3h;后将反应液加入1000.00g温度为5~15℃的水中,保温搅拌1~2h后过滤,滤饼用水漂洗后,在50℃干燥得321.27g棕色固体,hplc纯度:99.5%,收率为92%。
[0043]
化合物4的制备
[0044][0045]
在10~20℃之间,依次向反应瓶中加入433.97g的13%的甲胺甲醇溶液、400.00g的甲醇和200.00g的化合物3,保温搅拌1~2h;在-15~0℃之间,分批加入11.45g硼氢化钠,加毕后保温搅拌1h;控温在-15~10℃之间,将反应液滴加至1.5l的2n的盐酸中,滴加完毕体系ph为3~4;控温在50℃下,减压浓缩至无馏份后,加入1500.00g甲苯,降温至30℃以下,滴加氢氧化钠溶液调节至体系ph=10~11;分液,有机相用水洗涤一次,得到化合物4的甲苯溶液,纯度95%,理论收率100%。
[0046]
化合物5的制备
[0047][0048]
20~30℃下,在上步所得化合物4甲苯溶液中滴入二碳酸二叔丁酯132.14g,控温反应2~3h,反应液经600ml ph=2的稀盐酸洗涤,分液,得到化合物5的溶液。
[0049]
化合物4的制备
[0050][0051]
30~40℃下,在上步所得化合物5的甲苯溶液中滴加302.73g浓盐酸,搅拌1~3h后,加入500g 20%的氢氧化钠溶液,搅拌调节碱性,分液,脱色,得到化合物4的甲苯溶液。
[0052]
化合物6的制备
[0053][0054]
在25~35℃之间,向上步所得的化合物4的甲苯溶液中加入750g化合物6富马酸的甲醇溶液,滴加过程中体系有白色固体析出,滴加完毕,保温搅拌1~3h,过滤,滤饼在50℃干燥得到白色固体;
[0055]
在上步所得白色固体中加入1kg的纯化水、1kg的甲醇,升温至65~70℃后,搅拌溶清后,0~10℃之间保温搅拌析晶1~2h,过滤。滤饼用甲醇水混合液漂洗一次,滤饼在45℃干燥,得到203.15g白色固体即化合物7富马酸伏诺拉生,纯度100%,单杂<0.10%,收率为72.75%。
[0056]
实施例2:
[0057]
化合物3的制备
[0058]
向500ml反应瓶中依次加入530.00g四氢呋喃、200.00g的化合物1、25.83g的dmap、150.29g的dipea,开启搅拌,控温在15~35℃之间,滴加206.52g的化合物2,约30min左右滴加完毕;在15~35℃之间,保温反应1~3h;后将反应液加入1000.00g温度为5~15℃的水中,保温搅拌1~2h后过滤,滤饼用水漂洗后,在50℃干燥得320.22g棕色固体,hplc纯度:99.3%,收率为91.7%。
[0059]
化合物4的制备
[0060]
在10~20℃之间,依次向反应瓶中加入433.97g的13%的甲胺乙醇溶液、400.00g
的乙醇和200.00g的化合物3,保温搅拌1~2h;在-15~0℃之间,分批加入513.28g醋酸硼氢化钠,加毕后保温搅拌1h;控温在-15~10℃之间,将反应液滴加至1.5l的2n的盐酸中,滴加完毕体系ph为3~4;控温在50℃下,减压浓缩至无馏份后,加入1500.00g二氯甲烷,降温至30℃以下,滴加氢氧化钠溶液调节至体系ph=10~11;分液,有机相用水洗涤一次,得到化合物4的二氯甲烷溶液,纯度95%,理论收率100%。
[0061]
化合物5的制备
[0062]
15~19℃下,在上步所得化合物4的二氯甲烷溶液中滴入二碳酸二叔丁酯279.93g,控温反应2~3h,反应液经600ml 5%柠檬酸水溶液洗涤,分液,得到化合物5的二氯甲烷溶液。
[0063]
化合物4的制备
[0064]
30~40℃下,在上步所得化合物5的二氯甲烷溶液中滴加207.11g三氟乙酸,搅拌1~3h后,加入300g 20%的氢氧化钠溶液,搅拌调节碱性,分液,脱色,得到化合物4的二氯甲烷溶液。
[0065]
化合物6的制备
[0066]
在25~35℃之间,向上步所得的化合物4的二氯甲烷溶液中加入750g化合物6富马酸的甲醇溶液,滴加过程中体系有白色固体析出,滴加完毕,保温搅拌1~3h,过滤,滤饼在50℃干燥得到白色固体;
[0067]
在上步所得白色固体中加入1kg的纯化水、1kg的甲醇,升温至65~70℃后,搅拌溶清后,0~10℃之间保温搅拌析晶1~2h,过滤。滤饼用甲醇水混合液漂洗一次,滤饼在45℃干燥,得到181.51g白色固体即化合物7富马酸伏诺拉生,纯度100%,单杂<0.10%,收率为65%。
[0068]
实施例3:
[0069]
化合物3的制备
[0070]
向500ml反应瓶中依次加入530.00g二氯甲烷、200.00g的化合物1、25.83g的dmap、123.26g的碳酸钠,开启搅拌,控温在15~35℃之间,滴加206.52g的化合物2,约30min左右滴加完毕;在15~35℃之间,保温反应1~3h;后将反应液加入1000.00g温度为5~15℃的水中,保温搅拌1~2h后过滤,滤饼用水漂洗后,在50℃干燥得319.27g棕色固体,hplc纯度:99.3%,收率为91.5%。
[0071]
化合物4的制备
[0072]
在10~20℃之间,依次向反应瓶中加入433.97g的13%的甲胺甲醇溶液、400.00g的四氢呋喃和200.00g的化合物3,保温搅拌1~2h;在-15~0℃之间,分批加入26.63g氰基硼氢化钠,加毕后保温搅拌1h;控温在-15~10℃之间,将反应液滴加至1.5l的2n的盐酸中,滴加完毕体系ph为3~4;控温在50℃下,减压浓缩至无馏份后,加入1500.00g甲苯,降温至30℃以下,滴加氢氧化钠溶液调节至体系ph=10~11;分液,有机相用水洗涤一次,得到化合物4的甲苯溶液,纯度95%,理论收率100%。
[0073]
化合物5的制备
[0074]
31~45℃下,在上步所得化合物4甲苯溶液中滴入二碳酸二叔丁酯132.14g,控温反应2~3h,反应液经600ml ph=2的稀盐酸洗涤,分液,得到化合物5的溶液。
[0075]
化合物4的制备
[0076]
30~40℃下,在上步所得化合物5的甲苯溶液中滴加552.48g 20%的氲丝氢甲醇溶液,搅拌1~3h后,加入500g 20%的氢氧化钠溶液,搅拌调节碱性,分液,脱色,得到化合物4的甲苯溶液。
[0077]
化合物6的制备
[0078]
在25~35℃之间,向上步所得的化合物4的二氯甲烷溶液中加入750g化合物6富马酸的甲醇溶液,滴加过程中体系有白色固体析出,滴加完毕,保温搅拌1~3h,过滤,滤饼在50℃干燥得到白色固体;
[0079]
在上步所得白色固体中加入1kg的纯化水、1kg的甲醇,升温至65~70℃后,搅拌溶清后,0~10℃之间保温搅拌析晶1~2h,过滤。滤饼用甲醇水混合液漂洗一次,滤饼在45℃干燥,得到189.89g白色固体即化合物7富马酸伏诺拉生,纯度100%,单杂<0.10%,收率为68%。
[0080]
实施例4
[0081]
取市售原研参比制剂富马酸伏诺拉生片,商品名“沃克”(批号511492-1)进行检测,测得其中的富马酸伏诺拉生纯度为99.72%,杂质ii和杂质iii的含量分别为0.04%和0.09%,具体见附图3。
[0082]
富马酸伏诺拉生片具体的检测方法
[0083]
(1)制备供试品溶液
[0084]
1)将富马酸伏诺拉生片转移至200ml容量瓶,加入120ml稀释剂乙腈(参见下表)。
[0085]
片剂规格药片数量(=x)10mg620mg3
[0086]
2)用机械摇床剧烈振摇容量瓶,直至所有药片完全崩解,振摇条件:摇床:recipro shaker sr-1(水平振摇型,taitec corporation,日本)或等当品;振摇速度:约200次/min;振幅:约40mm。
[0087]
3)用稀释剂精确稀释至200ml,混合均匀。
[0088]
4)将此溶液通过孔径为0.45μm的膜滤器过滤。
[0089]
5)丢弃最初2ml或更多滤液,将后续滤液作为供试品溶液用于试验。
[0090]
注释:
[0091]
a)如果振摇30分钟后没有完全崩解,那么用超声处理或振动溶液,直至所有药片完全崩解。
[0092]
b)避光制备供试品溶液(如,在琥珀色玻璃瓶中,或在黄光下)
[0093]
c)当避光于大约25℃储存时(如hplc自动进样器),标准溶液可以使用72小时。
[0094]
d)如果经过论证,可以使用不同的振摇条件。
[0095]
(2)检测条件
[0096]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。