1.本发明属于农药技术领域,涉及砜吡草唑,具体涉及一种砜吡草唑稳定态晶型、制备方法及应用。
背景技术:
2.砜吡草唑(wo2002/062770)是由日本组合化学株式会社开发,并与日本庵原化学公司联合实现产业化的芽前土壤处理剂,属超长链脂肪酸延长合成酶抑制剂类除草剂,化学名称为[3-[(5-二氟甲氧基-1-甲基-3-三氟甲基吡唑-4-基)-甲基黄酰基]-4,5-二氢-5,5-二甲基-1,2-恶唑],结构如式1所示。其作用机制为通过抑制超长链脂肪酸延长合成酶vlcfae,从而破坏杂草幼苗分生组织与胚芽鞘,具有活性高、用量少、杀草谱广、安全性好等优点。
[0003][0004]
砜吡草唑于2011年在澳大利亚、2012年在美国及加拿大获得登记。国内由上海群力化工有限公司于2019年1月进行原药登记(pd20190017、pd20190059)。其在国内的合成专利(cn1257895c)保护期至2022年。国内有关的专利申请大部分涉及砜吡草唑与其它原药的组合物及应用,比如授权专利cn109588416a、cn109730080a、cn106070244a,分别采用敌草隆、甲酰胺基嘧磺隆、环吡氟草酮与砜吡草唑组合使用。专利cn111393427a及cn111574511a对砜吡草唑的合成工艺进行优化并申请保护。虽然砜吡草唑有上述诸多性能优势和市场优势,但对其晶型没有全面深入的拓展性研究。
[0005]
对原药晶型进行转换并择优筛选,有助于获得性能优异且适合市场推广的理想晶型产品。化学物质在结晶时受各种因素影响,分子内或分子间键合方式一般会发生改变,使分子或原子在晶格空间排列方式发生变化,形成不同的晶型,特定方法适用于特定一种晶型的成核和生长。在材料结晶过程施加不同条件,比如溶液的冷却、挥发、加压、对材料加热导致的熔融、升华等,都是普遍采用的晶型转换方法。其中溶液结晶法因具有易产生多晶型物、温度条件温和、操作简便、易于市场推广等优势,是目前最普遍的晶型转换方法。晶型差异通常对原药的物化性质和药效有较大影响,同一材料的不同晶型在稳定性、溶解度、熔点、溶出速度等方面通常会有显著差异,稳定型晶体分子堆叠紧凑规则,热熵小,熔点高,具有较高的化学稳定性,这对原药的安全运输与储存具有重要意义,有利于产品的推广应用。相反,亚稳态晶型或无定形态的原药在常规条件下易于转化成其它多晶型,导致产品性状
发生不可预测和不可控的变化,甚至产生结块、附聚、吸潮变质等现象,非常不利于产品的推广应用。
[0006]
参照公开的方法制备砜吡草唑,反应后多采用向反应体系中加水或不良溶剂的方法使砜吡草唑固体从体系中析出,由于体系含有杂质,且加水或不良溶剂的量和温度没有特定工艺,析出的白色固体纯度不够高,外观为粉末状,无特定晶型。另外这种细粉末的性状使其在过滤时易堵塞漏斗,导致过滤步骤耗时较多。另外其在干燥过程易聚集成块,干燥效果不均匀,影响干燥效率。储存时容易附着在器皿内壁并有吸潮现象。采用传统的溶剂重结晶法虽然对提升纯度有一定作用,但其步骤一般是分先后加入良溶剂和不良溶剂的溶解和析晶过程,需依赖物料溶解的现象不断调整溶剂量与加热温度,析晶工艺不固定,析出的晶型种类波动较大,且多为混合晶型,不利于产品的质量控制和剂型设计。因此,对砜吡草唑进行晶型变体转换,获得其稳定晶型,对提高产品纯度、优化生产工艺和储存稳定性、丰富制剂类型等方面,均具有重要意义。
技术实现要素:
[0007]
针对现有技术存在的不足,本发明的目的在于,提供一种砜吡草唑亚稳态、稳定态晶型、制备方法及应用,解决现有技术中的砜吡草唑难以获得稳定钛单一晶型的技术问题。
[0008]
为了解决上述技术问题,本发明采用如下技术方案予以实现:
[0009]
一种砜吡草唑稳定态晶型的制备方法,该方法包括以下步骤:
[0010]
步骤一,砜吡草唑在第一复配溶剂中搅拌加热,使固体溶解完全,降温冷却析晶,过滤干燥后为白色细针状晶体,即为亚稳态细针状晶型a;
[0011]
步骤二,亚稳态细针状晶型a与第二复配溶剂混合于压力釜中,惰性气体氛围下加压,保温保压,保持反应釜密封进行反应,降温冷却析晶,泄压后过滤干燥,为半透明柱状晶体b,即为稳定态柱状晶型b;
[0012]
所述的第一复配溶剂和第二复配溶剂均由溶剂i与溶剂ii按1:5~5:1的体积比组合而成,其中:
[0013]
所述的溶剂i选自酮类、酯类、卤代烷类和醚类中的一种或多种混合溶剂;
[0014]
所述的溶剂ii选自水、醇类、烷烃类和芳烃类中的一种或多种混合溶剂。
[0015]
本发明还具有如下技术特征:
[0016]
优选的,所述的溶剂i选自丁酮、乙酸正丁酯、三氯甲烷和四氢呋喃中的一种或多种混合溶剂;所述的溶剂ii选自甲醇或甲醇/水组合、正庚烷和甲苯中的一种或多种混合溶剂。
[0017]
进一步优选的,所述的第一复配溶剂为丁酮与甲醇/水的混合溶剂,丁酮与甲醇/水的体积比为(2.8~3):(0.7:0.3);所述的第二复配溶剂为乙酸正丁酯与正庚烷的混合溶剂,且乙酸正丁酯与正庚烷的体积比为(1.5~3):1。
[0018]
更具体的,该方法包括以下步骤:
[0019]
步骤一,砜吡草唑与复配溶剂按照质量体积比为10~200g/l混合,砜吡草唑在复配溶剂中搅拌加热,加热温度为30~110℃,加热10~30分钟使固体溶解完全,温度降至25~0℃冷却析晶,析晶时间1~48小时,过滤干燥后为白色细针状晶体,即为亚稳态细针状晶型a;
[0020]
步骤二,亚稳态细针状晶型a与复配溶剂以质量体积比20~350g/l混合于压力釜中,惰性气体氛围下加压至0.5~3mpa,在压力釜中的加热温度为50℃~150℃,加热时间为0.5~2.5小时,保持反应釜密封,温度降至25~0℃冷却析晶,析晶时间1~72小时,泄压后过滤干燥,为半透明柱状晶体b,即为稳定态柱状晶型b。
[0021]
本发明还保护一种砜吡草唑稳定态晶型,该砜吡草唑稳定态晶型采用如上所述的砜吡草唑稳定态晶型的制备方法制得,即稳定态柱状晶型b。
[0022]
所述的砜吡草唑稳定态晶型的分子式为c
12h14
f5n3o4s,所述的砜吡草唑稳定态晶型具有下列键长和键角:
[0023]
[0024][0025]
本发明还保护如上所述的砜吡草唑稳定态晶型用于除草剂中的应用。
[0026]
本发明还保护一种砜吡草唑亚稳态晶型的制备方法,与如上所述的步骤一相同。即制得亚稳态细针状晶型a。
[0027]
本发明还保护如上所述的砜吡草唑亚稳态晶型的制备方法制得的砜吡草唑亚稳态晶型用于除草剂中的应用。
[0028]
本发明与现有技术相比,具有如下技术效果:
[0029]
(ⅰ)本发明克服了传统溶剂重结晶法工艺不稳定、晶型单一性差等缺点,通过优选两种或多种溶剂混合成特定比例的复配溶剂,固定了溶剂组成,其中白色细针状晶型a的获得是在常压下对一定量砜吡草唑在复配溶剂中进行简单的升温溶解和降温析晶过程,避免
了传统重结晶法在加热过程中频繁调节溶剂配比和加热温度;晶型b的获得是在加压升温和增加过饱和度的情况下,以复配溶剂对晶型a进行转晶处理,获得半透明柱状晶型b。
[0030]
(ⅱ)两种晶型与制备工艺中反应液析出的白色粉末状固体相比,纯度高、分散性好、晶型单一且确定、实现了两种特定晶型变体的转换,其中经高温、高压、高过饱和度处理的晶型b为稳定态晶型,因而稳定性更好,吸湿性更低,便于储存,由晶型b制备的单剂型或复配剂型的可湿性粉剂、水分散粒剂等固体制剂稳定性佳、药效好,对砜吡草唑的工业生产和推广应用具有很强的实用价值。
[0031]
(ⅲ)砜吡草唑晶型b制备的单剂型和复配剂型,对田间杂草的整体防治效果均较好,其中与异丙隆组合的复配剂型获得协同增效的整体防效,水分散粒剂也获得最佳的整体防效。
附图说明
[0032]
图1为晶型a的x射线粉末衍射图。
[0033]
图2为晶型b的x射线粉末衍射图。
[0034]
图3为晶型a和晶型b的x射线粉末衍射对比图。
[0035]
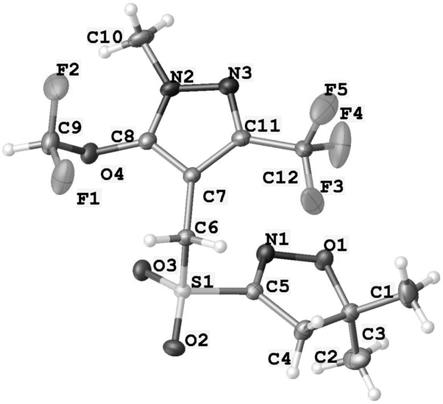
图4为晶型b的单晶x射线衍射结构图。
[0036]
图5为晶型b的单晶x射线衍射晶胞结构示意图。
[0037]
图6为晶型a和晶型b的热重分析图。
[0038]
图7为晶型a和晶型b的红外光谱图。
[0039]
图8为晶型a和晶型b的红外光谱局部放大图。
[0040]
图9为晶型a在氘代氯仿中的核磁共振氢谱图。
[0041]
图10为晶型b在氘代氯仿中的核磁共振氢谱图。
[0042]
以下结合实施例对本发明的具体内容作进一步详细解释说明。
具体实施方式
[0043]
需要说明的是,本发明中的所有原料,如无特殊说明,全部均采用现有技术中已知的原料。
[0044]
本发明的技术构思为:首先根据溶剂及溶质性质优选制备复配溶剂,然后对一定质量体积比的砜吡草唑固体和复配溶剂在常压下搅拌加热,溶解后冷却析晶;在加压条件下,增加上述已经呈现单一晶型的砜吡草唑与复配溶剂质量比,加热一定时间后,在一定压力下冷却析晶,在高的压力、温度、以及过饱和度多重转晶因素的作用下,得到外观不同的产物,经x射线粉末衍射分析、单晶x射线结构分析、红外光谱分析、热分析,确认砜吡草唑晶型发生转换,由亚稳态变为稳定态晶型。
[0045]
本发明中,作为初始原料的砜吡草唑选自非晶型或含有非晶型的砜吡草唑、或非单一晶型砜吡草唑的混合物。
[0046]
本发明中,草唑亚稳态细针状晶型a,即晶型a;稳定态柱状晶型b,即晶型b。
[0047]
遵从上述技术方案,以下给出本发明的具体实施例,需要说明的是本发明并不局限于以下具体实施例,凡在本技术技术方案基础上做的等同变换均落入本发明的保护范围。
[0048]
对比例1:
[0049]
将20g粉末状非单一晶型的砜吡草唑加入盛有100ml丁酮的250ml三口圆底烧瓶中,安装回流冷凝管,然后在80℃温度下以磁子剧烈搅拌(800转/分钟)至溶液清亮,再逐步加入甲醇/水=0.7:0.3的混合液,直至溶液发生浑浊,滴加几滴丁酮继续加热搅拌刚好能恢复清亮,得到热过饱和溶液。将该过饱和度溶液在搅拌的情况下降温,约1小时降至室温,并逐渐析出白色固体,重结晶过程持续6小时后,过滤干燥,得到含白色粉末状和细针状的混合固体18.2g,液相色谱含量98.0%,收率89%,为非特定单一晶型。
[0050]
对比例2:
[0051]
将29.7g砜吡草唑的a晶型加入盛有110ml乙酸正丁酯/正庚烷(1.8:1)复配溶剂的500ml压力釜中,以氮气置换釜内空气3次,密封釜后继续充氮气至压力达到2.4mpa,在不搅拌及室温情况下,保持该状态20小时,泄压后过滤干燥,得到夹杂少量柱状的白色细针状固体28.2g,液相色谱含量96.8%,收率92%,为未溶完全的原料及部分析晶产物的混合物。
[0052]
对比例3:
[0053]
将20g粉末状非单一晶型的砜吡草唑加入盛有110ml丙酮/(甲醇/水)=2.9:(0.7:0.3)复配溶剂的250ml三口圆底烧瓶中,安装回流管,在56℃温度下以磁子剧烈搅拌(800转/分钟)20分钟至溶液清亮,停止加热,以冰水浴降温冷却至3℃左右,析出较少白色固体,持续搅拌12小时,过滤结晶,得到少量白色细针状晶体11.6g,液相色谱含量99.4%,收率58%,部分砜吡草唑无法析出。
[0054]
对比例4:
[0055]
将29.7g粉末状非单一晶型的砜吡草唑加入盛有110ml乙酸正丁酯/正庚烷体积比=1.8:1的复配溶剂的500ml压力釜中,以氮气置换釜内空气3次,密封釜后继续充氮气至压力达到2.1mpa,在不搅拌的情况下,升温至120℃,此时压力达到2.4mpa,保持该状态2小时,停止加热。压力釜自然降温冷却至25℃左右,并持续该状态18小时以进行析晶,泄压后过滤干燥,得到含少量柱状晶体、其余大部分为白色粉末至细针状的固体27.8g,液相色谱含量99.2%,收率93%,所得砜吡草唑晶型状态较复杂,为非特定单一晶型。
[0056]
实施例1:
[0057]
本实施例给出一种砜吡草唑稳定态晶型的制备方法,其特征在于,该方法包括以下步骤:
[0058]
将20g粉末状非单一晶型的砜吡草唑加入盛有110ml丁酮/(甲醇/水)体积比=2.9:(0.7:0.3)的复配溶剂的250ml三口圆底烧瓶中,安装回流管,在65℃温度下以磁子剧烈搅拌(800转/分钟)20分钟至溶液清亮,停止加热,自然降温冷却至25℃左右,逐渐析出白色固体,室温下持续搅拌6小时,过滤结晶,得到白色细针状晶体17.8g,液相色谱含量99.0%,收率89%,其熔点经测试为134.2℃,经x射线粉末衍射法测试为特定晶型,如图1所示,称作晶型a。其热重分析如图6所示,其傅里叶变化红外光谱如图7及图8所示,其核磁共振氢谱如图9所示。
[0059]
将29.7g砜吡草唑的a晶型加入盛有110ml乙酸正丁酯/正庚烷体积比=1.8:1的复配溶剂的500ml压力釜中,以氮气置换釜内空气3次,密封釜后继续充氮气至压力达到2.1mpa,在不搅拌的情况下,升温至120℃,此时压力达到2.4mpa,保持该状态2小时,停止加热。压力釜自然降温冷却至25℃左右,并持续该状态18小时以进行析晶,泄压后过滤干燥,
得到半透明柱状晶体27.8g,液相色谱含量99.2%,收率93%,其熔点经测试为131.7℃,经x射线粉末衍射法测试为特定单一晶型,如图2所示,称作晶型b。其单晶x射线衍射结构解析如图4和图5所示,热重分析如图6所示,其傅里叶变化红外光谱如图7及图8所示,其核磁共振氢谱如图10所示。
[0060]
表征与性能测试:
[0061]
本发明中的晶型a的x射线粉末衍射图如图1所示,2θ值在5.0
°±
0.2
°
、10.0
°±
0.2
°
、15.0
°±
0.2
°
、20.0
°±
0.2
°
、22.0
°±
0.2
°
、22.8
°±
0.2
°
、25.2
°±
0.2
°
、30.2
°±
0.2
°
、31.6
°±
0.2
°
处有特征峰。
[0062]
本发明中的稳定态晶型b,的x射线粉末衍射图如图2所示,2θ值在5.1
°±
0.2
°
、7.6
°±
0.2
°
、10.0
°±
0.2
°
、11.2
°±
0.2
°
、15.0
°±
0.2
°
、17.8
°±
0.2
°
、20.0
°±
0.2
°
、22.4
°±
0.2
°
、25.6
°±
0.2
°
、27.0
°±
0.2
°
、28.4
°±
0.2
°
、30.2
°±
0.2
°
、31.6
°±
0.2
°
、32.4
°±
0.2
°
、34.6
°±
0.2
°
、35.6
°±
0.2
°
处有特征峰。
[0063]
图3为晶型a和晶型b的x射线粉末衍射对比图。
[0064]
如图4所示,采用单晶x射线衍射法对晶型b的晶体结构进行解析,明确表征晶型b晶体结构的重要参数,即所述的砜吡草唑稳定态晶型具有下列键长和键角:
[0065]
[0066][0067]
如图5所示,由晶型b的晶体结构及晶胞所获得的的晶体结构参数为:
[0068][0069][0070]
采用热重分析对两种不同外观的晶体进行测试(图6),晶型a在升温至298
℃
前随温度上升过程有约7%的重量损失,298
℃
之后随温度上升重量损失加快。晶型b在升温至302
℃
前随温度上升过程有约3%的重量损失,302
℃
之后随温度上升重量损失加快。晶型b的加速热损失起始温度高于晶型a。且在加速热损失起始温度以下,晶型b在同样加热温度下的质量损失小于晶型a,晶型b为稳定态晶型,晶型a为亚稳态晶型。
[0071]
采用傅里叶变换红外光谱法(ft-ir)对两种不同外观的晶体进行测试(图7、图8),晶型a的红外特征峰与晶型b的红外特征峰在波数为440cm-1
、708cm-1
、768cm-1
、906cm-1
、924cm-1
、1033cm-1
、1301cm-1
、1376cm-1
、1382cm-1
、1462cm-1
、1580cm-1
、2952cm-1
、3078cm-1
、3400cm-1
处的峰形和峰强度上有差异,说明两种晶型分子内的键合方式发生变化。
[0072]
采用核磁共振氢谱(1hnmr)对两种晶型的溶液进行结构表征(图9、图10),可见两者在氘代氯仿溶液中结构一致。氢谱解析如下,晶型a溶液:1h nmr(cdcl3,500mhz)δ(ppm):6.62-6.90(t,1h),4.53(s,2h),3.81(s,3h),3.03(s,2h),1.45(s,6h)。晶型b溶液:1h nmr(cdcl3,500mhz)δ(ppm):6.69-6.97(t,1h),4.60(s,2h),3.88(s,3h),3.10(s,2h),1.52(s,6h)。说明二者在溶液状态下晶型特征消失,结构相同,且核磁氢谱不含其它杂峰,说明两种方法获得的晶型a与晶型b均为高纯度产品。
[0073]
采用熔点仪在室内环境下对两种晶型的熔点进行测试,晶型a的熔点为131.7
℃
,晶型b的熔点为134.2
℃
,晶型b的熔点高。
[0074]
对晶型a和晶型b的稳定性和吸湿性进行测试,晶型b的外观、晶型、和质量在一定的温度和湿度范围内均无明显变化。
[0075]
对晶型b和晶型a配置了可湿性粉剂和水分散粒剂单剂型,并与异丙隆组合成可湿性粉剂和水分散粒剂复配剂型,这些剂型,尤其是基于晶型b的剂型在常温、低温、高温环境
下均表现出很好的稳定性。对这些基于晶型b的剂型进行了田间药效试验,采用芽前土壤喷雾处理法,对牛筋草、马唐、稗草、狗尾草、藜、反枝苋等的整体防效均在91%以上,最高超过94%,防杂草效果佳。
[0076]
实施例2:
[0077]
本实施例给出一种砜吡草唑稳定态晶型的制备方法,其特征在于,该方法包括以下步骤:
[0078]
将22g粉末状非单一晶型的砜吡草唑加入盛有110ml丁酮/正庚烷(3.2:1)复配溶剂的250ml三口圆底烧瓶中,安装回流管,在98℃温度下以磁子剧烈搅拌(800转/分钟)30分钟至溶液清亮,停止加热,冷却至室温,逐渐析出白色固体,持续搅拌8小时,过滤结晶,得到白色细针状晶体18.4g,液相色谱含量99.1%,收率83%,经表征晶型同实施例1,为晶型a。
[0079]
将31.9g砜吡草唑的a晶型加入盛有110ml四氢呋喃/甲苯(3:1)复配溶剂的500ml压力釜中,以氮气置换釜内空气3次,密封釜后继续充氮气至压力达到1.8mpa,在不搅拌的情况下,升温至120℃,此时压力达到2.3mpa,保持该状态2小时,停止加热。压力釜自然降温冷却至25℃后,转入0℃低温浴槽保温,并持续该状态12小时以进行析晶,泄压后过滤干燥,得到半透明柱状晶体28.6g,液相色谱含量99.1%,收率89%,经表征晶型同实施例2,为晶型b。
[0080]
实施例3:
[0081]
本实施例给出一种砜吡草唑稳定态晶型的制备方法,其特征在于,该方法包括以下步骤:
[0082]
将19.8g粉末状非单一晶型的砜吡草唑加入盛有110ml三氯甲烷/甲醇(1.8:1)复配溶剂的250ml三口圆底烧瓶中,安装回流管,在65℃温度下以磁子剧烈搅拌(800转/分钟)30分钟至溶液清亮,停止加热,冷却至室温,逐渐析出白色固体,持续搅拌20小时,过滤结晶,得到白色细针状晶体17.4g,液相色谱含量99.2%,收率87%,经表征晶型同实施例1,为晶型a。
[0083]
将30.9g砜吡草唑的a晶型加入盛有110ml三氯甲烷/甲苯(1.8:1)复配溶剂的500ml压力釜中,以氮气置换釜内空气3次,密封釜后继续充氮气至压力达到1.8mpa,在不搅拌的情况下,升温至100℃,此时压力达到2.2mpa,保持该状态2小时,停止加热。压力釜自然降温冷却至25℃后,转入0℃低温浴槽保温,并持续该状态12小时以进行析晶,泄压后过滤干燥,得到半透明柱状晶体27.7g,液相色谱含量99.2%,收率89%,经表征晶型同实施例2,为晶型b。
[0084]
实施例4:
[0085]
本实施例给出一种砜吡草唑稳定态晶型的制备方法,其特征在于,该方法包括以下步骤:
[0086]
将20g粉末状非单一晶型的砜吡草唑加入盛有110ml四氢呋喃/甲苯/甲醇(1.9:0.3:0.7)复配溶剂的250ml三口圆底烧瓶中,安装回流管,在66℃温度下以磁子剧烈搅拌(800转/分钟)20分钟至溶液清亮,停止加热,以冰水浴降温冷却至5℃左右,逐渐析出白色固体,持续搅拌6小时,过滤结晶,得到白色细针状晶体17.1g,液相色谱含量99.2%,收率85%,经表征晶型同实施例1,为晶型a。
[0087]
将29.7g砜吡草唑的a晶型加入盛有110ml四氢呋喃/三氯甲烷/正庚烷(1:0.5:1)
复配溶剂的500ml压力釜中,以氮气置换釜内空气3次,密封釜后继续充氮气至压力达到2.0mpa,在不搅拌的情况下,升温至110℃,此时压力达到2.4mpa,保持该状态2小时,停止加热。压力釜自然降温冷却至25℃左右,并持续该状态18小时以进行析晶,泄压后过滤干燥,得到半透明柱状晶体24.2g,液相色谱含量99.3%,收率81%,经表征晶型同实施例2,为晶型b。
[0088]
实施例5:
[0089]
本实施例给出一种砜吡草唑稳定态晶型用于除草剂中的应用。其中,晶型a和晶型b均采用实施例1中制得的晶型a和晶型b。
[0090]
一、晶型稳定性实验:
[0091]
实验方法:将砜吡草唑晶型a和晶型b固体在不同温度下储存14天,复测其晶型变化情况。
[0092]
实验数据:
[0093][0094]
实验结论:砜吡草唑晶型b稳定,常规储存无明显转晶现象;晶型a较稳定,温度较高时有一定量的无定形转化现象发生。
[0095]
二、晶型吸湿性实验:
[0096]
实验方法:砜吡草唑晶型a和晶型b固体分别取100克,在不同湿度下储存7天,复测其质量及外观变化情况。
[0097]
实验数据:
[0098][0099]
实验结论:砜吡草唑晶型b在设定湿度和温度范围内无明显吸湿性;晶型a在较低湿度和温度范围内无明显吸湿性,但随湿度和温度提升到较高水平时,表现出轻微的吸潮板结现象。
[0100]
三、剂型配置及田间药效实验:
[0101]
由上述实验一及实验二结论可见,砜吡草唑晶型b在晶体结构及储存稳定性方面具有显著优势,在固体剂型的配置方面同样具有明显的优势。
[0102]
本发明获得的砜吡草唑晶型b,可单独或与其它除草剂复配组合,作为有效活性成分,再与农业上常用可接受的助剂制备成适应剂型,优选剂型为可湿性粉剂、水分散粒剂等。
[0103]
1、30%砜吡草唑可湿性粉剂配方如下:
[0104]
物料折百质量比(%)砜吡草唑晶型b/晶型a40烷基芳基聚氧乙烯基醚10十二烷基苯磺酸钠9硬脂酸镁6硫酸铵10硅藻土补足至100
[0105]
该剂型制备方法为:将上述物料在混合机中混匀,经气流粉碎机粉碎,并进一步在混合机中混合,再通过600目标准筛干燥后,得到砜吡草唑可湿性粉剂。
[0106]
对所得可湿性粉剂产品的理化性质进行测试,常温下是在室温下密封放置14天后测试含量,冷储存是在0℃下密封放置7天后测试含量,热储存是在54℃下密封放置14天后测试含量,稳定性好,可满足使用要求,结果见下表:
[0107][0108]
2、40%砜吡草唑/异丙隆(20:20)可湿性粉剂配方如下:
[0109][0110][0111]
该剂型制备方法为:将上述物料在混合机中混匀,经气流粉碎机粉碎,并进一步在混合机中混合,再通过600目标准筛干燥后,得到砜吡草唑/异丙隆可湿性粉剂。
[0112]
对所得复配可湿性粉剂产品的理化性质进行测试,常温下是在室温下密封放置14天后测试含量,冷储存是在0℃下密封放置7天后测试含量,热储存是在54℃下密封放置14天后测试含量,稳定性好,可满足使用要求,结果见下表:
[0113][0114]
3、30%砜吡草唑水分散粒剂配方如下:
[0115]
物料折百质量比(%)砜吡草唑晶型b/晶型a30
木质素磺酸钠15膨润土15聚二甲基硅氧烷0.5高岭土补足至100
[0116]
该剂型制备方法为:将上述物料在混合机中混匀,经气流粉碎机粉碎,并进一步在混合机中混合,再经600目标准筛后加入捏合机中捏合成可塑性物料,将此物料加入挤压造粒机中挤压造粒,干燥后得到砜吡草唑水分散粒剂。
[0117]
对所得水分散粒剂产品的理化性质进行测试,常温下是在室温下密封放置14天后测试含量,冷储存是在0℃下密封放置7天后测试含量,热储存是在54℃下密封放置14天后测试含量,稳定性好,可满足使用要求,结果见下表:
[0118][0119]
4、40%砜吡草唑/异丙隆(20:20)水分散粒剂配方如下:
[0120]
物料折百质量比(%)砜吡草唑晶型b/晶型a20异丙隆20茶枯粉7硫酸铵5可溶性淀粉5磷酸氢二钠4膨润土补足至100
[0121]
该剂型制备方法为:将上述物料在混合机中混匀,经气流粉碎机粉碎,并进一步在混合机中混合,再经600目标准筛后加入捏合机中捏合成可塑性物料,将此物料加入挤压造粒机中挤压造粒,干燥后得到砜吡草唑/异丙隆水分散粒剂。
[0122]
对所得水分散粒剂产品的理化性质进行测试,常温下是在室温下密封放置14天后测试含量,冷储存是在0℃下密封放置7天后测试含量,热储存是在54℃下密封放置14天后测试含量,稳定性好,可满足使用要求,尤其由晶型b获得的制剂更佳,结果见下表:
[0123][0124]
5、田间杂草药效试验
[0125]
针对稳定性好的晶型b制剂进行田间杂草药效试验。
[0126]
所选试验田的田间发生杂草主要有牛筋草、马唐、稗草、狗尾草、藜、反枝苋等。
[0127]
试验方法:在杂草发芽前进行手动喷雾处理土壤,兑水量40公斤/亩,小区面积50平方米,每处理重复4次,施药后45天调查防除效果。其中杂草株防效(%)=(清水对照区杂草株数—药剂处理区杂草株数)/清水对照区杂草株数。防效结果如下表:
[0128]
实施组剂型用量(ga.i./ha)整体防效(%)40%砜吡草唑可湿性粉剂18091.250%砜吡草唑 异丙隆(20 30)可湿性粉剂22593.330%砜吡草唑水分散粒剂20091.940%砜吡草唑 异丙隆(20 20)水分散粒剂22594.2
[0129]
由药效试验结果可见,由砜吡草唑晶型b制备的单剂型和复配剂型,对田间杂草的整体防治效果均较好,其中与异丙隆组合的复配剂型获得协同增效的整体防效,水分散粒剂也获得最佳的整体防效。
[0130]
上述应用例只为说明砜吡草唑稳定晶型的实际应用价值,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。