1.本发明属于药物化学领域,具体涉及一种含杂环串联类化合物及其制备方法与应用。
背景技术:
2.shp2是一个在体内广泛存在的非受体型蛋白酪氨酸磷酸酶,具有两个n末端src同源性2结构域(n-sh2和c-sh2)、催化结构域(ptp)和c末端尾部。这两个sh2结构域控制shp2的亚细胞定位和功能调节。作为血小板源性生长因子(pdgf)、表皮生长因子(egf)、成纤维细胞因子(fgf)、白细胞介素-3(il-3)、白血病抑制因子(lif)及α-干扰素(inf-α)等生长因子的下游信号分子,shp2参与ras/mapk通路、pi3k/akt通路、jak/stat通路、jnk通路等在内的多条信号通路。因此,发现和寻找具有较好成药性的shp2抑制剂逐渐成为工业界和学术界的一大热点研究领域。
技术实现要素:
3.本发明需要解决的技术问题之一是提供一类末端为杂环串联的全新shp2抑制剂,以解决目前shp2抑制剂结构骨架单一等问题。
4.解决上述技术问题的方案如下:
5.一种如通式i所示的化合物,及其药学上可接受的盐、对映异构体、非对映异构体、互变异构体、溶剂化物、多晶型物或前药
[0006][0007]
r1、r2和r3分别独立地为氢、卤素、氨基;
[0008]
x为n或ch;
[0009]
l为键、o或s;
[0010]
独立地是单键或双键:
[0011]
当为双键时,y1为n,y2为ch;
[0012]
当为单键时,y1为c=o,y2为nra或crbrc,其中ra,rb和rc分别独立地为氢、c1-c3的烷基、c1-c3的羟烷基、c1-c3的烷氧基;
[0013]
r4为或其中,n=0或1;
[0014]
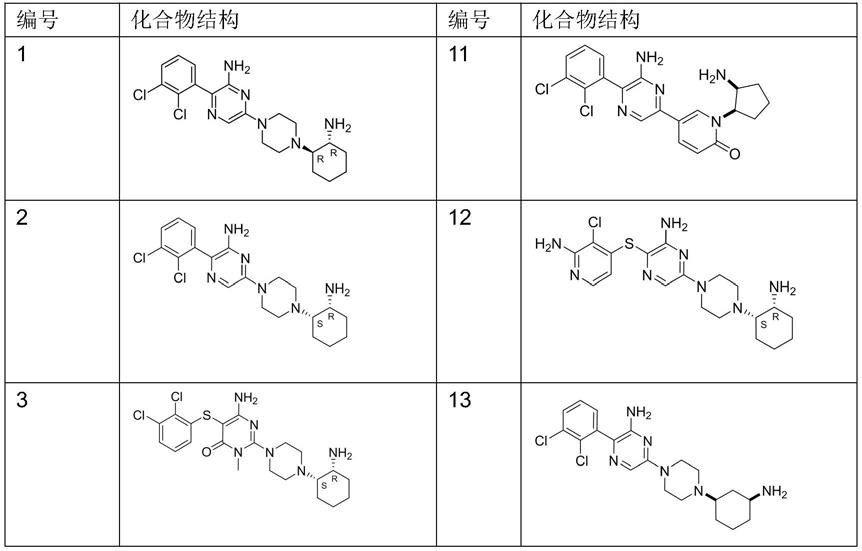
所述的化合物,其特征在于所述化合物为如下结构式中任意一个:
[0015]
[0016][0017]
一种药物组合物,其特征在于含有所述的杂环串联类化合物和药学可接受的辅料。
[0018]
所述的药物组合物,其特征在于药物组合物制成片剂、胶囊剂、注射液或冻干粉剂。
[0019]
所述的杂环串联类化合物、所述的药物组合物在制备治疗抗肿瘤药物、作为抗肿瘤药物的前药或作为抗肿瘤药物的中间体中应用。
[0020]
有益效果
[0021]
本发明首次公开一系列含有新骨架的含杂环串联类化合物,该类化合物为shp2抑制剂,该类化合物具有一定抗肿瘤活性,为后续开发抗肿瘤药物提供支持。
具体实施方式
[0022]
中间体3-氯-4-碘-2-吡啶胺(a1)的合成:
[0023][0024]
步骤一:2-氟-3-氯-4-碘吡啶(a1-2)的合成:
[0025]-78℃下,将正丁基锂(38ml,1.25eq)缓慢滴加到a1-1(10.00g,76.3mmol)的thf(75ml)溶液中。反应1h后缓慢滴加i2的thf(30ml)溶液。反应30min后监测。监测反应完全后,滴加饱和na2so3水溶液淬灭,浓缩除thf,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析分离得化合物a1-2(7.76g,收率41%)。1h nmr(300mhz,cdcl3):δ7.77(dd,j=5.2,0.9hz,1h),7.67(d,j=5.2hz,1h).esi-ms m/z:257.9[m h]
.
[0026]
步骤二:3-氯-4-碘-2-吡啶胺(a1)的合成
[0027]
将nh3·
h2o(38ml)缓慢滴加到a1-2(7.56g,29.4mmol)的dmso(38ml)溶液中。加毕,80℃封管反应过夜。监测反应完全后,将反应体系倒入水(200ml)中搅拌30min,抽滤干燥得
化合物a1(6.79g,收率91%)。1h nmr(300mhz,cdcl3)δ7.57(d,j=5.2hz,1h),7.12(d,j=5.2hz,1h),5.05(s,2h).esi-ms m/z:254.9[m h]
.
[0028]
中间体6-氯-3-(2,3-二氯丙基)吡嗪-2-氨基叔丁酯(b1)的合成:
[0029][0030]
步骤一:6-氯-3-(2,3-二氯苯基)吡嗪-2-胺(b1-2)的合成
[0031]
将化合物b1-1(5.00g,24.0mmol,1.0eq)、2,3-二氯苯硼酸(5.04g,26.4mmol,1.1eq)、pd(dppf)cl2(350.1mg,2mol%)和k3po4(10.18g,48.0mmol,2.0eq)置于200ml单口瓶中,将体系抽真空置换氮气,加入1,4-二氧六环(54ml)和水(6ml)于120℃油浴中反应过夜,监测至原料转化完全。硅藻土过滤,滤液浓缩,加入30ml乙酸乙酯萃取,饱和氯化钠水溶液水洗3次,浓缩,柱层析纯化,得化合物b1-2(5.94g,收率91%)。1h nmr(300mhz,cdcl3):δ8.03(s,1h),7.60(dd,j=7.6,2.0hz,1h),7.36(dd,j=7.7,7.6hz,1h),7.32(dd,j=7.6,2.0hz,1h),4.65(s,2h).esi-ms m/z:274.1[m h]
.
[0032]
步骤二:6-氯-3-(2,3-二氯苯基)吡嗪-2-氨基叔丁酯(b1)的合成
[0033]
将化合物b1-2(5.30g,19.4mmol)和dmap(118.0mg,0.97mmol,0.05eq)置于200ml单口瓶中,加入二氯甲烷(50ml),在0℃加入二碳酸二叔丁酯,加毕,移至室温反应2h,监测至原料转化完全。饱和氯化钠水溶液水洗3次(15ml
×
3),有机相浓缩,柱层析纯化,得化合物b1(7.50g,收率82%)。1h nmr(300mhz,cdcl3):δ8.68(s,1h),7.57(d,j=7.5hz,1h),7.46
–
7.24(m,2h),1.37(s,18h).esi-ms m/z:474.1[m h]
.中间体b2的合成:
[0034][0035]
步骤一:6-氨基-3-甲基嘧啶-2,4(1h,3h)-二酮(b2-2)的合成
[0036]
将浓h2so4(0.1ml)缓慢滴加到4-氨基-2,6-二羟基嘧啶(5.18g,40.8mmol)的hmds(25ml)溶液中。130℃反应6h后,浓缩除hmds。加入dmf(25ml),碘甲烷(8.5ml,3.5eq),室温反应过夜。监测反应完全后,滴加nahco3(a.q.)至无气泡产生,抽滤,滤饼水洗,干燥得化合物b2-2(3.60g,收率63%)。1h nmr(300mhz,dmso-d6):δ10.43(s,1h),6.23(brs,2h),4.59(s,1h),3.00(s,3h).
[0037]
步骤二:6-氨基-5-碘-3-甲基嘧啶-2,4(1h,3h)-二酮(b2)的合成
[0038]
将化合物b2-2(1.95g,13.83mmol)溶于dmf(14ml)和acoh(44ml)中,加入nis(3.73g,1.2eq)。室温反应2h,监测反应完全后,抽滤,滤饼水洗干燥,得中间体b2(3.06g,收率83%)。1h nmr(300mhz,dmso-d6)δ10.66(s,1h),6.26(s,2h),3.10(s,3h).
[0039]
中间体b3-a,b3-b的合成
[0040][0041]
步骤一:
[0042]
0℃下,将nbs(23.3g)和ts-oh(2.05g)加入到dcm(60ml)溶液中,搅拌10h,将化合物b3-1(10g)的dcm(120ml)溶液加入到体系中,50℃回流过夜,监测反应完全后,萃取,柱层析纯化,得油状化合物b3-2(17.35g,crude)。
[0043]
步骤二:
[0044]
0℃下,将氢化钠(4.14g,103.4mmol)加入到化合物b3-2(16.76g,103.4mmol)的dmf(150ml)溶液中,室温反应1h后,0℃下缓慢加(15g,86.2mmol),室温过夜。监测反应完全后,萃取,柱层析纯化,得化合物b3-3(4.43g,15%over2 steps)。1h nmr(300mhz,cdcl3)δ7.37(dd,j=9.7,2.7hz,1h),7.26(d,j=2.7hz,1h),6.48(d,j=9.7hz,1h),4.47
–
4.37(m,1h),2.71
–
2.55(m,1h),2.49
–
2.20(m,4h),2.00
–
1.81(m,1h).
[0045]
步骤三:
[0046]
室温下,将ti(opr-i)4(8.88g,9.25ml)加入到化合物b3-3(4g,15.6mmol)的氨的甲醇溶液(20ml)中,反应4h后,缓慢加入硼氢化钠(886mg,1.5eq)室温反应3h。监测反应完全后,萃取,柱层析纯化,得先洗脱产物b3-a(1.04g,26%),1h nmr(300mhz,cdcl3)δ7.61(d,j=2.7hz,1h),7.37(dd,j=9.6,2.7hz,1h),6.50(d,j=9.6hz,1h),5.07
–
4.88(m,1h),3.84
–
3.71(m,1h),2.24
–
1.44(m,2h).后洗脱产物b3-b(1.25g,31%),1h nmr(300mhz,cdcl3)δ7.46(d,j=2.7hz,1h),7.33(dd,j=9.6,2.7hz,1h),6.50(d,j=9.6hz,1h),5.07
–
4.97(m,1h),3.60
–
3.49(m,1h),2.32
–
2.16(m,2h),2.03
–
1.68(m,4h).
[0047]
中间体6-溴-3-(4-(4-丁氧基羰基)哌嗪-1-基)吡嗪-2-羧酸甲酯(b4)的合成:
[0048][0049]
将3,6-二溴吡嗪-2-甲酸甲酯(500.0mg,1.69mmol)、n-boc哌嗪(346.2mg,1.1eq)及dipea(1.09g,5eq)溶于乙腈(8ml),室温搅拌过夜。监测反应完全后,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析分离得化合物b4(498.6mg,收率74%)。1h nmr(300mhz,cdcl3)δ8.24(s,1h),3.97(s,3h),3.59
–
3.41(m,8h),1.48(s,9h).esi-ms m/z:498.2[m h]
.
[0050]
中间体5-氯吡嗪-2-硫醇钠(b5)的合成:
[0051][0052]
步骤一:3-((5-氯吡嗪-2-基)硫代)丙酸乙酯(b5-1)的合成
[0053]
将3-巯基丙酸乙酯(4.45ml,1.05eq)缓慢滴加到2,5-二氯吡嗪(5.00g,33.6mmol)和k2co3(4.64g,1eq)的dmf(42ml)溶液中。室温反应4h。监测反应完全后,乙酸乙酯稀释,饱
和食盐水水洗5次,有机相浓缩,柱层析分离得化合物b5-1(7.78g,收率94%)。1h nmr(300mhz,cdcl3)δ8.39(d,j=1.5hz,1h),8.22(d,j=1.5hz,1h),4.17(q,j=7.2hz,2h),3.42(t,j=7.0hz,2h),2.75(t,j=7.0hz,2h),1.27(t,j=7.2hz,3h).esi-ms m/z:247.0[m h]
.
[0054]
步骤二:5-氯吡嗪-2-硫醇钠(b5)的合成
[0055]
将乙醇钠(2.24g,1.1eq)缓慢滴加到b5-1(7.38g,30mmol)的thf(100ml)溶液中。室温反应2h。监测反应完全后,加正己烷(100ml)打浆抽滤,固体干燥得化合物b5(5.13g,粗品)。1h nmr(300mhz,dmso-d6)δ7.82(d,j=1.3hz,1h),7.77(d,j=1.3hz,1h).esi-ms m/z:147.0[m h]
.
[0056]
中间体c1的合成:
[0057][0058]
0℃下,将甘氨酸甲酯盐酸盐(1eq),hatu(1.1eq),dipea(1.5eq)加到c1-1(5.00g,21.62mmol)的dcm溶液中,室温反应5h。tlc板监测反应完全后,ch2cl2稀释,饱和食盐水水洗5次,有机相浓缩,柱层析分离得化合物c1-2(6.4g,收率98%)。esi-ms m/z:303.1[m h]
.
[0059]
将hcl(30ml,2m in etoac)加入到化合物c1-2(80mg,0.11mmol)的etoac(20ml)溶液中,室温过夜。tlc板监测至原料转化完全,抽滤除滤液,固体干燥得化合物c1-3(4.7g,90%)。esi-ms m/z:203.1[m h]
.
[0060]
将dipea(3eq)加到化合物c1-3(4.70g,23.3mmol)的甲醇(70ml)溶液中,35℃反应过夜。tlc板监测至原料转化完全后,将反应液浓缩,加入etoac(20ml)打浆得化合物c1-4(5.8g,crude)。esi-ms m/z:171.1[m h]
.
[0061]
将lialh4(4eq)加到化合物c1-4(5.80g,34.1mmol)的thf(70ml)溶液中,70℃反应过夜。tlc板监测至原料转化完全后,饱和nh4cl(5ml)缓慢加入。抽滤除固体,滤液浓缩得化合物c1-5(3.3g,crude)。esi-ms m/z:143.1[m h]
.
[0062]
将cbz-cl(0.6eq),naoh(20ml,2m in etoac)加到c1-5(3.3g,23.2mmol)的thf(20ml)溶液中,室温反应过夜。tlc板监测反应完全后,加入etoac(15ml)和水(10ml)萃取分液。合并有机层并用盐水(3
×
10ml)洗涤,有机相经na2so4干燥,减压蒸馏除去溶剂。残余物通过柱色谱纯化,得到化合物c1-6(330.0mg,15%over 3steps)。esi-ms m/z:277.1[m h]
.
[0063]
0℃下,将邻苯二甲酰亚胺(1.5eq)和三苯基膦(1.5eq)加入到化合物c1-6(315.2mg,1.14mmol)的thf(4ml)中,氮气保护后,加入dead(1.5eq)的thf(2ml)溶液,加毕室温反应过夜。监测反应完全后,有机相浓缩,柱层析纯化,得化合物c1-7(1.07g,crude)。esi-ms m/z:406.2[m h]
.
[0064]
将pd/c(10%wt)加入到化合物c1-7(1.07g,2.64mmol)的甲醇(5ml)溶液中。将反应体系置换氢气后,室温搅拌过夜。监测反应完全后,抽滤,浓缩,柱层析纯化,得化合物c1(45.6mg,18%over 2steps)。1h nmr(300mhz,chloroform-d)δ7.86
–
7.70(m,4h),4.94
–
4.79(m,1h),3.40
–
2.11(m,11h).esi-ms m/z:272.1[m h]
.
[0065]
中间体c2的合成:
[0066]
参照中间体c1的合成方法,得到c2。1h nmr(300mhz,chloroform-d)δ7.87
–
7.68(m,4h),4.98
–
4.84(m,1h),3.41
–
2.49(m,9h),2.23
–
2.13(m,1h),1.95
–
1.80(m,1h).esi-ms m/z:272.1[m h]
。
[0067]
实施例1:
[0068][0069]
步骤一:
[0070]
将苄溴(1.1eq)和碳酸钾(3eq)加入到化合物1-1(3g,16.8mmol)的乙腈(50ml)溶液中,80℃反应3h。tlc板监测至原料转化完全,萃取,柱层析纯化,得化合物1-2(3.74g,96%)。esi-ms m/z:232.1[m h]
.
[0071]
步骤二:
[0072]
将碳酸氢钠(3.6eq)和((1r,2r)-2-氨基环己基)氨基甲酸叔丁酯(1.1eq)加入到化合物1-2(1g,4.33mmol)的乙醇(15ml)溶液中,氮气保护,90℃回流5h。监测反应完全后抽滤浓缩,柱层析纯化,得化合物1-3(880.1mg,55%)。esi-ms m/z:374.3[m h]
.
[0073]
步骤三:
[0074]
将pd/c(10%wt)和水合肼(2eq)加入到化合物1-3(700mg,1.87mmol)的乙醇(10ml)溶液中,氮气保护,80℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物1-4(420mg,85%)。esi-ms m/z:284.2[m h]
.
[0075]
步骤四:
[0076]
将化合物b1(0.66eq)和碳酸铯(2.5eq)加入到化合物1-4(180mg,0.63mmol)的dmso(3ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物1-5(181.2mg,60%)。esi-ms m/z:721.3[m h]
.
[0077]
步骤五:
[0078]
将hcl(3ml,2m in etoac)加入到化合物1-5(80mg,0.11mmol)的etoac(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用
etoac萃水相,合并有机相,浓缩得化合物1(33mg,71%)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.4,2.2hz,1h),7.37
–
7.25(m,2h),4.24(s,2h),3.69
–
3.47(m,4h),2.87
–
2.65(m,3h),2.59
–
2.46(m,2h),2.23
–
2.11(m,1h),2.06
–
1.94(m,4h),1.88
–
1.76(m,2h),1.30
–
1.07(m,4h).esi-ms m/z:421.2[m h]
。
[0079]
实施例2:
[0080][0081]
步骤一:
[0082]
将boc酸酐(3.16g,14.5mmol)加入到化合物2-1(2.0g,13.2mmol)的dcm(30mmol)溶液中,滴加三乙胺(2.9g,29mmol),室温过夜。监测反应完全后,dcm萃取,无水硫酸钠干燥,浓缩得化合物2-2(3.14g,粗品)。esi-ms m/z:216.2[m h]
.
[0083]
步骤二:
[0084]
将邻苯二甲酰亚胺(2.98g,20.25mmol)和三苯基膦(5.3g,20.25mmol)加入到化合物2-2(2.9g,13.5mmol)的thf(40ml)中,氮气保护,0℃下加入dead(3.5g,20.25mmol)的thf,15min后移至室温,反应过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物2-3(3.15g,68%)。esi-ms m/z:345.2[m h]
.
[0085]
步骤三:
[0086]
将水合肼(585mg,11.7mmol)加入到化合物2-3(3.1g,9.01mmol)的甲苯(20ml)溶液中,80℃回流过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物2-4(1.09g,56%)。esi-ms m/z:215.2[m h]
.
[0087]
步骤四:
[0088]
将碳酸氢钠(1.79g,21.30mmol)和化合物1-2(1.24g,4.63mmol)加入到化合物2-4(1.09g,5.09mmol)的乙醇(15ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物2-5(1.18g,62%)。esi-ms m/z:374.3[m h]
.
[0089]
步骤五:
[0090]
将pd/c(200mg,20%wt)和水合肼(295mg,5.9mmol)加入到化合物2-5(1.1g,2.95mmol)的乙醇(10ml)溶液中,氮气保护,80℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物2-6(0.99g,粗品)。esi-ms m/z:284.2[m h]
.
[0091]
步骤六:
[0092]
将化合物b1(300mg,0.634mmol)和碳酸铯(310mg,0.95mmol)加入到化合物2-6(197mg,0.697mmol)的dmso(3ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合
物2-7(215mg,47%)。esi-ms m/z:721.2[m h]
.
[0093]
步骤七:
[0094]
将hcl(3ml,2m in etoac)加入到化合物2-7(215mg,mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物2(91.2mg,73%)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.4,2.3hz,1h),7.35
–
7.28(m,2h),4.24(s,2h),3.58(t,j=5.1hz,4h),3.41
–
3.35(m,1h),2.76
–
2.54(m,4h),2.12
–
2.01(m,1h),1.88
–
1.70(m,3h),1.56
–
1.33(m,4h),1.31
–
1.12(m,1h).esi-ms m/z:421.2[m h]
。
[0095]
实施例3:
[0096][0097]
步骤一:
[0098]
将化合物2-6(334.4mg,1.18mmol,)、bop(990.7mg,2.24mmol)和dbu(1.2g,7.84mmol)溶于dmf(3ml)中,室温反应过夜,tlc板监测至原料转化完全,加etoac(15ml),有机相用饱和氯化钠溶液(5ml)洗几次,柱层析纯化,得化合物3-2(247mg,42%)。esi-ms m/z:533.2[m h]
.
[0099]
步骤二:
[0100]
将2,3-二氯苯硫醇(107.4mg,0.6mmol)、cui(1.5mg,0.007mmol)、tmeda(1.9mg,0.016mmol)和磷酸钾(254.7mg,1.2mmol)溶于化合物3-2(212.4mg,0.4mmol)的dioxane(2.5ml)溶液中,氮气保护,100℃反应过夜。tlc板监测至原料转化完全,抽滤除去固体,柱层析纯化,得化合物3-3(68.4mg,29%)。esi-ms m/z:583.2[m h]
.
[0101]
步骤三:
[0102]
将hcl(3ml,2m in etoac)加入到化合物3-3(53.7mg,0.09mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物3(43.1mg,97%)。1h nmr(300mhz,cdcl3)δ7.19(dd,j=7.8hz,1.2hz,1h),7.02(t,j=7.8hz,1h),6.81(dd,j=7.8hz,1.2hz,1h),5.30(s,2h),3.44(s,3h),3.40
–
3.23(m,5h),2.79
–
2.57(m,4h),2.15
–
2.05(m,1h),1.89
–
1.69(m,3h),1.51
–
1.33(m,4h),1.30
–
1.16(m,1h).esi-ms m/z:406.2[m h]
.esi-ms m/z:483.1[m h]
。
[0103]
实施例4:
[0104][0105]
步骤一:
[0106]
将boc酸酐(3.6g,16.3mmol)和盐酸(1ml)加入到化合物4-1(1.53g,13.6mmol)的甲醇(23ml)溶液中,滴加4m的hcl异丙醇溶液1.7ml,室温过夜。监测反应完全后,萃取,无水硫酸钠干燥,柱层析纯化得化合物4-2(415mg,14%)。esi-ms m/z:201.2[m h]
.
[0107]
步骤二:
[0108]
将碳酸氢钠(697.2mg,8.3mmol)和化合物1-2(483mg,1.8mmol)加入到化合物4-2(380mg,1.8mmol)的乙醇(5.2ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物4-3(506.1mg,65%)。esi-ms m/z:374.3[m h]
.
[0109]
步骤三:
[0110]
将pd/c(96mg,20%wt)和水合肼(130mg,2.6mmol)加入到化合物4-3(480mg,1.3mmol)的乙醇(4ml)溶液中,氮气保护,80℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物4-4(267.7mg,73%)。esi-ms m/z:284.2[m h]
.
[0111]
步骤四:
[0112]
将化合物b1(200mg,0.42mmol)和碳酸铯(205.4mg,0.63mmol)加入到化合物4-4(133.5mg,0.42mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物4-5(129.9mg,43%)。esi-ms m/z:721.3[m h]
.
[0113]
步骤五:
[0114]
将hcl(3ml,2m in etoac)加入到化合物4-5(106.4mg,0.15mmol)的etoac(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物4(55mg,89%)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.4,2.2hz,1h),7.36
–
7.28(m,2h),4.23(s,2h),3.70
–
3.48(m,4h),2.88
–
2.76(m,2h),2.75
–
2.64(m,1h),2.59
–
2.47(m,2h),2.23
–
2.11(m,1h),2.06
–
1.96(m,1h),1.74
–
1.63(m,1h),1.29
–
1.05(m,4h).esi-ms m/z:421.2[m h]
。
[0115]
实施例5:
[0116][0117]
步骤一:
[0118]
将boc酸酐(1.5g,6.82mmol)加入到化合物5-1(1.0g,6.22mmol)的dcm溶液中,滴加三乙胺(752.9mg,7.44mmol),室温过夜。监测反应完全后,etoac萃取,无水硫酸钠干燥,浓缩得化合物5-2(1.9g,粗品)。esi-ms m/z:250.1[m h]
.
[0119]
步骤二:
[0120]
将邻苯二甲酰亚胺(1.59g,10.8mmol)和三苯基膦(2.84g,10,83mmol)加入到化合物5-2(1.8g,7.22mmol)的thf(18ml)中,氮气保护,0℃下加入diad(2.19g,10.83mmol)的thf(4ml),15min后移至室温,反应过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物5-3(1.8g,67%)。esi-ms m/z:379.2[m h]
.
[0121]
步骤三:
[0122]
将水合肼(480mg,9.6mmol)加入到化合物5-3(1.8g,4.8mmol)的甲苯(14ml)溶液中,80℃回流过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物5-4(637mg,54%)。esi-ms m/z:249.2[m h]
.
[0123]
步骤四:
[0124]
将碳酸氢钠(1.04g,12.42mmol)和化合物1-2(725.2mg,2.7mmol)加入到化合物5-4(663mg,2.7mmol)的乙醇(10ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物5-5(768.3mg,70%)。esi-ms m/z:407.2[m h]
.
[0125]
步骤五:
[0126]
将pd/c(143.04mg,20%wt)和水合肼(575mg,11.5mmol)加入到化合物5-5的乙醇(8ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物5-6(559.7mg,98%)。esi-ms m/z:317.2[m h]
.
[0127]
步骤六:
[0128]
将化合物b1(146.2mg,0.46mmol)和碳酸铯(205.3mg,0.63mmol)加入到化合物5-6(200mg,0.42mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物5-7(184.4mg,58%)。esi-ms m/z:755.3[m h]
.
[0129]
步骤七:
[0130]
将hcl(3ml,2m in etoac)加入到化合物5-7(100mg,0.132mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物5(47.8mg,80%)。1h nmr(300mhz,cdcl3)δ7.61(s,
1h),7.51(dd,j=7.4,2.2hz,1h),7.42
–
7.37(m,1h),7.36
–
7.28(m,2h),7.26
–
7.19(m,3h),4.31(d,j=5.1hz,1h),4.26(s,2h),3.67(t,j=4.7hz,4h),3.04
–
2.86(m,3h),2.83
–
2.58(m,4h).esi-ms m/z:455.1[m h]
。
[0131]
实施例6:
[0132][0133]
步骤一:
[0134]
将boc酸酐(1.1eq)加入到化合物6-1(0.96g,6.98mmol)的dcm(15ml)溶液中,滴加三乙胺(2.2eq),室温过夜。监测反应完全后,etoac萃取,无水硫酸钠干燥,浓缩得化合物6-2(1.6g,粗品)。esi-ms m/z:202.1[m h]
.
[0135]
步骤二:
[0136]
将邻苯二甲酰亚胺(1.5eq)和三苯基膦(1.5eq)加入到化合物6-2(1.4g,6.9mmol)的thf(20ml)中,氮气保护,0℃下加入dead(1.5eq)的thf(2ml),15min后移至室温,反应过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物6-3(1.73g,75%)。esi-ms m/z:331.2[m h]
.
[0137]
步骤三:
[0138]
将水合肼(1.3eq)加入到化合物6-3(1.7g,5.15mmol)的甲苯(15ml)溶液中,80℃回流过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物6-4(1.005g,97%)。esi-ms m/z:201.2[m h]
.
[0139]
步骤四:6-5的合成
[0140]
将碳酸氢钠(4.6eq)和化合物1-2(1eq)加入到化合物6-4(1g,5mmol)的乙醇(10ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物6-5(1.07g,59%)。esi-ms m/z:360.3[m h]
.
[0141]
步骤五:
[0142]
将pd/c(20%wt)和水合肼(2eq)加入到化合物6-5(1g,2.78mmol)的乙醇(9ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物6-6(550mg,73%)。esi-ms m/z:270.2[m h]
.
[0143]
步骤六:
[0144]
将化合物b1(0.83eq)和碳酸铯(1.5eq)加入到化合物6-6(135mg,0.5o mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物6-7(207mg,84%)。esi-ms m/z:707.2[m h]
.
[0145]
步骤七:
[0146]
将hcl(3ml,2m in etoac)加入到化合物6-7(100mg,0.132mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物6(42.6mg,79%)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.3,2.3hz,1h),7.36
–
7.28(m,2h),4.26(s,2h),3.61(t,j=5.0hz,4h),3.43(t,j=4.3hz,1h),2.72
–
2.50(m,4h),2.39
–
2.26(m,1h),1.97
–
1.55(m,6h).esi-ms m/z:407.2[m h]
。
[0147]
实施例7:
[0148][0149]
步骤一:
[0150]
将boc酸酐(1.1eq)加入到化合物7-1(1g,8.68mmol)的dcm(15ml)溶液中,滴加三乙胺(1.2eq),室温过夜。监测反应完全后,etoac萃取,无水硫酸钠干燥,浓缩得化合物7-2(2.1g,粗品)。esi-ms m/z:216.2[m h]
.
[0151]
步骤二:
[0152]
将邻苯二甲酰亚胺(1.5eq)和三苯基膦(1.5eq)加入到化合物7-2(1.4g,6.51mmol)的thf(20ml)中,氮气保护,0℃下加入dead(1.5eq)的thf(2ml),15min后移至室温,反应过夜。监测反应完全后有机相浓缩,柱层析纯化,得化合物7-3(1.26g,56%)。esi-ms m/z:345.2[m h]
.
[0153]
步骤三:
[0154]
将水合肼(3eq)加入到化合物7-3(1.26g,3.64mmol)的甲苯(15ml)溶液中,80℃回流2h。监测反应完全后有机相浓缩,柱层析纯化,得化合物7-4(757mg,99%)。esi-ms m/z:215.2[m h]
.
[0155]
步骤四:
[0156]
将碳酸氢钠(4.6eq)和化合物1-2(1.1eq)加入到化合物7-4(757mg,3.6mmol)的乙醇(10ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物7-5(0.85g,74%)。esi-ms m/z:374.2[m h]
.
[0157]
步骤五:
[0158]
将pd/c(20%wt)和水合肼(2eq)加入到化合物7-5(0.75g,2mmol)的乙醇(9ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物7-6(620mg,crude)。esi-ms m/z:284.2[m h]
.
[0159]
步骤六:
[0160]
将化合物b1(1eq)和碳酸铯(1.5eq)加入到化合物7-6(143mg,0.5mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物7-7(116.4mg,49%)。esi-ms m/z:721.3[m h]
.
[0161]
步骤七:
[0162]
将hcl(3ml,2m in etoac)加入到化合物7-7(116.4mg,0.25mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物7(42.6mg)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.4,2.2hz,1h),7.36
–
7.28(m,2h),4.25(s,2h),3.70
–
3.53(m,4h),3.45
–
3.39(m,1h),2.76
–
2.56(m,4h),2.14
–
2.05(m,1h),1.94
–
1.72(m,4h),1.54
–
1.40(m,3h),1.32
–
1.18(m,1h).esi-ms m/z:421.2[m h]
。
[0163]
实例8:
[0164][0165]
步骤一:
[0166]
将boc酸酐(6.3g,28.93mmol)和加入到化合物8-1(3g,26.3mmol)的甲醇(50ml)溶液中,滴加三乙胺3.98g,室温过夜。监测反应完全后,萃取,无水硫酸钠干燥,柱层析纯化得化合物8-2(1.79g,32%)。esi-ms m/z:215.2[m h]
.
[0167]
步骤二:
[0168]
将碳酸氢钠(1.3g,15.37mmol)和化合物1-2(1g,4.27mmol)加入到化合物8-2(1g,4.70mmol)的乙醇(10ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物8-3(1.32g,80%)。esi-ms m/z:374.2[m h]
[0169]
步骤三:
[0170]
将pd/c(165mg,20%wt)和水合肼(187mg,3.74mmol)加入到化合物8-3(700mg,1.87mmol)的乙醇(10ml)溶液中,氮气保护,80℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物8-4(320mg,60%)。esi-ms m/z:284.2[m h]
.
[0171]
步骤四:
[0172]
将化合物b1(209mg,0.44mmol)和碳酸铯(215mg,0.66mmol)加入到化合物8-4(150mg,0.53mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物8-5(136mg,43%)。esi-ms m/z:721.3[m h]
.
[0173]
步骤五:
[0174]
将hcl(3ml,2m in etoac)加入到化合物8-5(136mg,0.15mmol)的etoac(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物8(56mg,89%)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.4,2.3hz,1h),7.36
–
7.28(m,2h),4.24(s,2h),3.58(t,j=5.1hz,4h),3.42
–
3.36(m,1h),2.75
–
2.54(m,4h),2.08(dt,j=11.9,3.4hz,1h),1.92
–
1.81(m,2h),1.58
–
1.14(m,6h).esi-ms m/z:421.2[m h]
。
[0175]
实施例9:
[0176][0177]
步骤一:
[0178]
将中间体a1(3.00g,11.8mmol)、中间体b5(1.99g,1eq)、pd2(dba)3(216.3mg,2mol%)、xantphos(273.2mg,4mol%)、dipea(3.04g,2eq)置于封管中,氮气保护后加入无水二氧六环(50ml)于100℃反应过夜。监测反应完全后,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析分离得化合物9-1(2.33g,收率72%)。1h nmr(300mhz,cdcl3):δ8.52(d,j=1.5hz,1h),8.36(d,j=1.5hz,1h),7.90(d,j=5.2hz,1h),6.58(d,j=5.2hz,1h),5.04(brs,2h).esi-ms m/z:273.0[m h]
.
[0179]
步骤二:
[0180]
将化合物9-1(100.0mg,0.37mmol)和化合物2-6(124.5mg,0.44mmol)溶于dipea:nmp(1:1,2ml)溶液中,95℃反应过夜,监测反应完全后,萃取,柱层析纯化,得化合物9-2(78.5mg,41%)。esi-ms m/z:520.2[m h]
.
[0181]
步骤三:
[0182]
将hcl(3ml,2m in etoac)加入到化合物9-2(65.1mg,0.13mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物9(20.2mg,38%)。1h nmr(300mhz,cdcl3)δ8.27(d,j=1.3hz,1h),8.20(d,j=1.3hz,1h),7.69(d,j=5.4hz,1h),6.01(d,j=5.4hz,1h),4.88(s,2h),3.71
–
3.64(m,4h),3.41
–
3.35(m,1h),2.77
–
2.59(m,4h),2.12
–
2.04(m,1h),1.87
–
1.76(m,3h),1.51
–
1.38(m,4h),1.26
–
1.17(m,1h).esi-ms m/z:420.2[m h]
。
[0183]
实例10:
[0184][0185]
步骤一:
[0186]
将boc酸酐(1.1eq)加入到化合物10-1(1g,3.89mmol)的dcm(15ml)溶液中,滴加三乙胺(1.2eq),室温过夜。监测反应完全后,etoac萃取,无水硫酸钠干燥,柱层析纯化,得化合物10-2(606.1mg,41%)。esi-ms m/z:357.1[m h]
.
[0187]
步骤二:
[0188]
将bpin(1.5eq),pd(dppf)cl2(5mol%)和ch3cook(2.5eq)加入到化合物10-2(300mg,0.84mmol)的无水二氧六环(3ml)液体中,90℃反应1.5h,监测反应完全后,萃取,柱层析纯化,得化合物10-3(141.2mg,42%)。esi-ms m/z:405.2[m h]
.
[0189]
步骤三:
[0190]
将pd(dppf)cl2(5mol%),na2co3(2eq)和中间体b1(1eq)加入到化合物10-3(141.2mg)的二氧六环和水的混合溶液中,90℃反应1.5h,监测反应完全后,萃取,柱层析纯化,得化合物10-4(106.9mg,43%)。esi-ms m/z:716.3[m h]
.
[0191]
步骤四:
[0192]
将hcl(3ml,2m in etoac)加入到化合物10-4(79mg)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物10(43.8mg,95%)。1h nmr(300mhz,cdcl3)δ8.36(d,j=2.4hz,1h),8.30(s,1h),7.97(dd,j=9.5,2.5hz,1h),7.62
–
7.55(m,1h),7.41
–
7.34(m,2h),6.68(d,j=9.5hz,1h),5.14
–
5.02(m,1h),4.64(s,2h),3.90
–
3.79(m,1h),2.33
–
2.16(m,2h),2.14
–
1.96(m,2h),1.73
–
1.52(m,2h).esi-ms m/z:416.1[m h]
。
[0193]
实例11:
[0194][0195]
步骤一:
[0196]
将boc酸酐(1.1eq)加入到化合物11-1(1.2g,4.7mmol)的dcm(15ml)溶液中,滴加三乙胺(1.2eq),室温过夜。监测反应完全后,etoac萃取,无水硫酸钠干燥,柱层析纯化,得化合物11-2(1.12mg,67%)。esi-ms m/z:357.1[m h]
.
[0197]
步骤二:
[0198]
将bpin(1.5eq),pd(dppf)cl2(5mol%)和ch3cook(2.5eq)加入到化合物11-2
(450mg,1.26mmol)的无水二氧六环(5ml)液体中,90℃反应1.5h,监测反应完全后,萃取,柱层析纯化,得化合物11-3(243.7mg,48%)。esi-ms m/z:405.2[m h]
.
[0199]
步骤三:
[0200]
将pd(dppf)cl2(5mol%),na2co3(2eq)和中间体b1(1eq)加入到化合物11-3(120mg,0.3mmol)的二氧六环和水的混合溶液中,90℃反应1.5h,监测反应完全后,萃取,柱层析纯化,得化合物11-4(91.3mg,41%)。esi-ms m/z:716.3[m h]
.
[0201]
步骤四:
[0202]
将hcl(3ml,2m in etoac)加入到化合物11-4(91.3mg)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物11(53.1mg,99%)。1h nmr(300mhz,cdcl3)δ8.28(s,1h),8.20(d,j=2.5hz,1h),7.93(dd,j=9.5,2.5hz,1h),7.64
–
7.55(m,1h),7.42
–
7.34(m,2h),6.69(d,j=9.5hz,1h),5.06
–
4.94(m,1h),4.59(s,2h),3.49(q,j=8.4hz,1h),2.39
–
2.12(m,2h),1.99
–
1.81(m,3h),1.57
–
1.49(m,1h).esi-ms m/z:416.1[m h]
。
[0203]
实施例12:
[0204][0205]
步骤一:
[0206]
将化合物2-6(131mg,0.46mmol)和碳酸钾(127.1mg,0.92mmol)加入到化合物12-1(132.5mg,0.46mmol)的nmp(2ml)溶液中,100℃反应过夜,监测反应完全后,萃取,柱层析纯化,得化合物12-2(79.1mg,38%)。esi-ms m/z:535.2[m h]
.
[0207]
步骤二:
[0208]
将hcl(3ml,2m in etoac)加入到化合物12-2(65mg,0.143mmol)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物12(43.2mg,85%)。1h nmr(300mhz,dmso-d6)δ7.63(d,j=5.4hz,1h),7.60(s,1h),6.27(s,2h),6.18(s,2h),5.72(d,j=5.4hz,1h),3.59
–
3.51(m,4h),3.34
–
3.24(m,1h),2.63
–
2.51(m,4h),2.01
–
1.92(m,1h),1.74
–
1.09(m,8h).esi-ms m/z:435.2[m h]
。
[0209]
实施例13:
[0210][0211]
步骤一:
[0212]
将(pho)2p(=o)n3(1.13eq),et3n(1.6eq),加入到化合物13-1(2g,8.22mmol)的甲苯(40ml)溶液中,室温反应3h。再加入苯甲醇(2eq),100℃反应过夜。监测反应完全后,萃
取,柱层析纯化,得化合物13-2(2.53g,88%)。esi-ms m/z:349.2[m h]
.
[0213]
步骤二:
[0214]
将pd/c(20%wt)加入到化合物13-2(1.5g,4.3mmol)的甲醇(20ml)溶液中,氢气条件下室温反应过夜,监测反应完全后,萃取,柱层析纯化,得化合物13-3(613.2mg,66%)。esi-ms m/z:215.2[m h]
.
[0215]
步骤三:
[0216]
将碳酸氢钠(1.1eq)和化合物1-2(1.1eq)加入到化合物13-3(600mg,2.3mmol)的乙醇(10ml)溶液中,氮气保护,85℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物13-4(730mg,80%)。esi-ms m/z:374.3[m h]
.
[0217]
步骤四:
[0218]
将pd/c(20%wt)和水合肼(4eq)加入到化合物13-4(690mg,1.85mmol)的乙醇(10ml)溶液中,氮气保护,80℃回流过夜。监测反应完全后抽滤浓缩,柱层析纯化,得化合物13-5(320mg,62%)。esi-ms m/z:284.2[m h]
。
[0219]
步骤五:
[0220]
将化合物b1(1eq)和碳酸铯(1.5eq)加入到化合物13-5(119.1mg,0.42mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(10ml),饱和氯化钠水溶液水洗多次(3ml
×
6),合并有机相,浓缩,柱层析纯化,得化合物13-6(170.3mg,56%)。esi-ms m/z:721.3[m h]
.
[0221]
步骤六:
[0222]
将hcl(3ml,2m in etoac)加入到化合物13-6(79mg)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物13(45mg,95%)。1h nmr(300mhz,cdcl3)δ7.59(s,1h),7.50(dd,j=7.4,2.3hz,1h),7.36
–
7.28(m,2h),4.24(s,2h),3.60(t,j=5.1hz,4h),2.81
–
2.61(m,5h),2.49
–
2.37(m,1h),2.11
–
2.02(m,1h),1.90
–
1.71(m,2h),1.40
–
0.92(m,5h).esi-ms m/z:421.2[m h]
。
[0223]
实施例14:
[0224][0225]
步骤一:
[0226]
将3,6-二溴吡嗪-2-甲酸甲酯(800.0mg,2.7mmol)、化合物2-6(1.1eq)及dipea
(5eq)溶于乙腈(12ml),室温搅拌过夜。监测反应完全后,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析分离得化合物14-1(1.22g,91%)。esi-ms m/z:498.2[m h]
.
[0227]
步骤二:
[0228]
将2-氨基-3-氯吡啶-4-硫代硫酸钠(1.5eq),pd2(dba)3(2mol%)、xantphos(6mol%)、dipea(3eq)加入到化合物14-1(1.1g,2.21mmol)的无水二氧六环(10ml)溶液中,氮气保护后于100℃反应过夜。监测反应完全后,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析分离得化合物14-2(1.0g,85%)。esi-ms m/z:578.2[m h]
.
[0229]
步骤三:
[0230]
将dibal-h(4eq)加入到化合物14-2(900mg,1.56mmol)的dcm(15ml)溶液中,室温反应过夜。监测反应完全后,乙酸乙酯萃取,无水硫酸钠干燥,浓缩,柱层析分离得化合物14-3(137mg,15%)。esi-ms m/z:550.2[m h]
.
[0231]
步骤四:
[0232]
将hcl(3ml,2m in etoac)加入到化合物14-3(65mg)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物14(12mg,23%)。1h nmr(300mhz,cdcl3)δ8.28(s,1h),7.70(d,j=5.4hz,1h),6.05(d,j=5.4hz,1h),5.01(s,2h),4.67(s,2h),3.51
–
3.30(m,4h),2.82
–
2.59(m,4h),2.20
–
2.09(m,1h),1.98
–
1.72(m,3h),1.59
–
1.14(m,6h).esi-ms m/z:450.2[m h]
。
[0233]
实施例15:
[0234][0235]
步骤一:
[0236]
将化合物9-1(64.1mg,0.23mmol)和化合物2-6(75.0mg,0.23mmol)溶于dipea:nmp(1:1,1.5ml)溶液中,95℃反应过夜,监测反应完全后,萃取,柱层析纯化,得化合物15-1(58.5mg,45%)。esi-ms m/z:554.2[m h]
.
[0237]
步骤二:
[0238]
将hcl(3ml,2m in etoac)加入到化合物15-1(65mg)的etoac(1ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物15(12mg,23%)。1h nmr(300mhz,cdcl3)δ8.29(d,j=1.3hz,1h),8.23(d,j=1.3hz,1h),7.70(d,j=5.4hz,1h),7.42
–
7.36(m,1h),7.26
–
7.20(m,3h),6.03(d,j=5.4hz,1h),4.89(s,2h),4.31(d,j=4.9hz,1h),3.82
–
3.73(m,4h),3.05
–
2.88(m,3h),2.86
–
2.76(m,2h),2.72
–
2.63(m,2h).esi-ms m/z:454.2[m h]
。
[0239]
实施例16:
[0240][0241]
步骤一:
[0242]
将化合物c1(95.1mg,0.35mmol)和碳酸铯(228.2mg,0.7mmol)加入到化合物b1(165.6mg,0.35mmol)的dmso(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,加入乙酸乙酯(30ml),饱和氯化钠水溶液水洗多次(6
×
3ml),合并有机相,浓缩,柱层析纯化,得化合物16-2(91.2mg,37%)。esi-ms m/z:709.2[m h]
.
[0243]
步骤二:
[0244]
将hcl(2ml,2m in etoac)加入到化合物16-2(91.2mg,0.13mmol)的etoac(2ml)溶液中,室温过夜。tlc板监测至原料转化完全,用水萃取,水相用饱和碳酸钠溶液调碱,再用etoac萃水相,合并有机相,浓缩得化合物16-3(58.2mg,粗品)。
[0245]
步骤三:
[0246]
将水合肼(3eq)加入到化合物16-3(58.2mg,0.12mmol)的乙醇(1ml)溶液中,60℃反应3h。监测反应完全后有机相浓缩,柱层析纯化,得化合物16(20.2mg,41%over 2steps)。esi-ms m/z:379.1[m h]
。
[0247]
实施例17:
[0248][0249]
步骤一:
[0250]
将化合物c1(45.6mg,0.17mmol)和碳酸钾(34.8mg,0.25mmol)加入到化合物12-1(53.9mg,0.19mmol)的nmp(2ml)溶液中,95℃反应过夜,监测反应完全后,萃取,柱层析纯化,得化合物17-1(30.0mg,34%)。1h nmr(300mhz,cdcl3)δ7.88
–
7.69(m,5h),7.66(s,1h),6.05(d,j=5.4hz,1h),4.92(brs,2h),4.84(brs,2h),4.49
–
4.41(m,1h),4.28
–
4.20(m,1h),3.46
–
3.39(m,1h),3.36
–
2.99(m,3h),2.78
–
2.68(m,1h),2.50
–
2.15(m,5h).esi-ms m/z:523.1[m h]
.
[0251]
步骤二:
[0252]
将水合肼(3eq)加入到化合物17-1(30.0mg,0.057mmol)的乙醇(1ml)溶液中,60℃反应3h。监测反应完全后有机相浓缩,柱层析纯化,得化合物16(14.8mg,66%)。1h nmr(300mhz,dmso-d6)δ7.67
–
7.62(m,2h),6.28(brs,2h),6.19(brs,2h),5.73(d,j=5.3hz,1h),4.49
–
4.40(m,1h),4.34
–
4.24(m,1h),3.57
–
3.33(m,1h),3.07
–
2.59(m,4h),2.33
–
1.86(m,4h),1.20
–
1.07(m,1h).esi-ms m/z:393.1[m h]
。
[0253]
实施例18的合成:
[0254]
参照实施例17的合成方法,得到实施例18。1h nmr(300mhz,chloroform-d)δ7.70(d,j=5.4hz,1h),7.64(s,1h),6.03(d,j=5.4hz,1h),4.87(brs,2h),4.84(brs,2h),4.47
–
4.39(m,1h),4.33
–
4.22(m,1h),3.73
–
3.63(m,1h),3.53
–
3.45(m,1h),3.13
–
3.00(m,2h),2.73
–
2.62(m,1h),2.41
–
2.27(m,2h),2.00
–
1.83(m,2h),1.64
–
1.55(m,1h).esi-ms m/z:393.1[m h]
。
[0255]
实施例19的合成:
[0256]
参照实施例17的合成方法,得到实施例19。h nmr(300mhz,dmso-d6)δ8.48(d,j=1.4hz,1h),8.31(d,j=1.4hz,1h),7.65(d,j=5.4hz,1h),6.36(brs,2h),5.80(d,j=5.4hz,1h),4.60
–
4.33(m,1h),4.41(d,j=12.9hz,1h),3.57
–
3.46(m,1h),3.12
–
2.69(m,4h),2.37
–
1.95(m,4h),1.29
–
1.18(m,1h).esi-ms m/z:378.1[m h]
。
[0257]
实施例20体外shp2酶水平活性测试
[0258]
对上述实施例中化合物的shp2酶水平活性进行测试,具体操作如下:
[0259]
4.1化合物配制
[0260]
化合物溶解在100%dmso中,配制成30mm储存液,于-20度冰箱避光保存。
[0261]
4.2shp2反应过程
[0262]
(1)配制1
×
retoacctionbuffer。
[0263]
(2)化合物浓度梯度的配制:受试化合物测试起始浓度为30μm,3倍稀释,10个浓度,单孔测试。在384source板中稀释成100倍终浓度的100%dmso溶液,用precision 3倍稀释化合物,10个浓度。使用分液器echo 550向目的板384板中转移250nl 100倍终浓度的化合物。正对照加入250nl dmso,负对照加入250nl 1mm shp099。
[0264]
(3)用1
×
retoacctionbuffer配制5倍终浓度的激活肽溶液,分别加入5μl到反应板中,
[0265]
1000rpm离心1min。
[0266]
(4)用1
×
retoacctionbuffer配制2.5倍终浓度的酶溶液,分别加入10μl到反应板中,1000rpm离心1min,室温孵育60分钟。
[0267]
(5)用1
×
retoacctionbuffer配制2.5倍终浓度的底物溶液,分别加入10μl到反应板中,1000rpm离心1min,室温孵育20分钟。
[0268]
(6)用ensight读取ex355/em460荧光数值
[0269]
4.3数据分析
[0270]
计算公式
[0271][0272]
其中:rfu:样品的荧光值;metoacn(nc):含10μm shp099的对照孔荧光值均值;metoacn(pc):阳性对照孔荧光值均值。
[0273]
拟合量效曲线以浓度的log值作为x轴,百分比抑制率为y轴,采用分析软件graphpad prism 5的log(inhibitor)vs.response-variable slope拟合量效曲线,从而得出各个化合物对酶活性的ic50值。
[0274]
计算公式是y=bottom (top-bottom)/(1 10^((logic50-x)*hillslope))
[0275]
具体结果如表所示:
[0276][0277][0278]
a《500nm,500nm≤b≤1000nm,c》1000nm。
[0279]
实验结论:以上数据显示,本发明实施例化合物对shp2磷酸酶具有变构抑制作用。
[0280]
化合物体外抗增殖活性
[0281]
实施例21细胞毒性实验
[0282]
1、实验步骤
[0283]
(1)pbs溶液进行高压灭菌,置于冰箱4℃保存。
[0284]
(2)称量胰蛋白酶和胰酶消化液,加入超纯水充分溶解,用微孔过滤器过滤得液体,置于冰箱-20℃保存。
[0285]
(3)分别称取培养基粉和nahco3,加入超纯水充分溶解,加入10%双抗,用微孔滤膜过滤得培养液,置于冰箱4℃保存,待使用前加入10%胎牛血清。
[0286]
(4)将nci-h358细胞从液氮罐中取出,立刻置于37摄氏度恒温水浴锅中,摇晃使其
融化,再将细胞倒入培养瓶中,加入培养液(含10%胎牛血清)稀释。将稀释后的培养基转入离心管中,1000r/min离心5分钟,舍弃上清液,再加入新鲜的培养基吹打混匀,移入培养瓶中培置于5%co2、37℃培养箱中培养。待细胞贴壁快铺满瓶底时开始进行传代,加入少量新鲜的培养基(含10%胎牛血清)终止消化,倒掉培养瓶中的液体,pbs洗两遍,加入新鲜培养基吹打混匀,均分到两个培养瓶中继续培养。
[0287]
(5)取对数期细胞,倒掉旧培养基,加入胰蛋白酶溶液消化3分钟,加入含10%胎牛血清的新鲜培养基终止消化,将溶液转移至离心管,1000r/min离心5分钟,舍弃上清液。加入培养基将其配制成细胞悬浊液,进行细胞计数。计数完成后,按照每孔5000-10000个细胞浓度将细胞植于96孔板中。将铺好细胞的96孔板置于37℃、5%co2培养箱中继续培养24h。用培养基将药物梯度稀释为90μmol/l,30μmol/l,10μmol/l,3.3μmol/l,1.1μmol/l,0.37μmol/l随后将它们加入到96孔板中,每孔100μl,每个浓度设置三个复孔。对照组加入相应浓度的含溶媒的培养基,调零孔加入相同体积的空白培养基,置于5%co2、37℃培养箱孵育3天,每两天更换一次培养基。每孔加入20μl mtt(5mg/ml),混合均匀后,于5%co2、37℃培养箱避光培养4h。将96孔板中的液体移除,每孔加入150μl dmso,置于微型振荡器上震荡,使底部的结晶完全溶解。随后将96孔板放入酶标仪中检测,于490nm处测定吸光度。
[0288]
2、数据处理
[0289]
绘制曲线并计算药物对细胞的抑制率及ic
50
。
[0290]
抑制率=[(对照组平均od值-实验组平均od值)/(对照组平均od值-空白对照组平均od值)]
×
100%。
[0291]
3、实验结果
[0292]
化合物对非小细胞肺癌细胞株nci-h358细胞的抑制活性如下:
[0293]
化合物编号抑制率(10μm)抑制率(50μm)241.8%53.1%339.5%60.0%558.2%60.2%1416.4%53.9%176.64%26.5%shp099-3%41%
[0294]
实验结论:以上数据显示,本发明实施例化合物对nci-h358细胞的增殖具有良好的抑制作用。相比较shp099而言,本发明实施例5具备新颖的结构和更优越的活性。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。