不受clsp阻碍物质影响的clsp衍生物和clsp活性的增强/保护剂
技术领域
1.本发明涉及:钙调素样皮肤蛋白(calmodulin-like skin protein:clsp)的衍生物,其具有抑制与阿尔茨海默病(ad)相关的神经细胞的功能障碍或神经细胞死亡的活性,并且不受阻碍物质(阻碍剂)对该活性的阻碍或抑制作用;clsp所具有的该活性(也称为“ad保护活性”、“抗ad活性”、“clsp活性”或“clsp的细胞毒性抑制活性”)的增强或保护剂,其由包含脂连蛋白的胶原同源区的多肽等构成;包含clsp或该clsp衍生物和该多肽等的融合蛋白;以及包含它们作为有效成分的药物组合物、特别是阿尔茨海默病治疗用药物组合物等。
背景技术:
2.阿尔茨海默病(ad)是引起痴呆的主要的神经变性疾病。ad的病因尚未充分阐明,而且用于ad的疾病修饰(疾病的预防和进展抑制)疗法还处于远未实用化的阶段(1-3)。
3.作为生理活性肽的人体肽(humanin)和clsp是针对由睫状神经营养因子受体α、wsx-1和gp130构成的异源三聚体人体肽受体(hthnr)的生理学激动剂(4-6)。它们经由hthnr在体外阻碍ad相关神经元细胞死亡(5、7)。另外,clsp的转基因过度表达保护其在ad模型小鼠中免于突触丧失和记忆丧失(8)。然而,人体肽的活性弱(50%有效浓度为1~10μm) (6、7),认为生物体内存在的人体肽的浓度不足以发挥神经保护效果(6、9)。
4.clsp主要在皮肤角化细胞中产生,在一部分外周组织的上皮细胞中也会产生少许(10-12)。通过clsp的腹腔内给药,小鼠的东莨菪碱诱发记忆障碍得到改善(13)。另外,在人脑脊液中存在足够量的clsp (14)。由这些实验事实推测:clsp从外周组织通过血液循环运输到达中枢神经系统(cns),透过血脑屏障进入神经组织(14)。由clsp中的第40~61位的22个氨基酸构成的序列即her (内源性人体肽同源区)是clsp活性不可缺少的(5),野生型clsp的活性比人体肽强105倍(50%有效浓度为10-100pm) (5)。另外,由测得的人脑脊液中的clsp的浓度(14)推测:cns中的clsp浓度是足以作为ad保护因子显示神经保护效果的浓度。由发表的这些见解(5、6、8、9、13和14)来看,体内的hthnr的核心激动剂很有可能是clsp而不是人体肽。另外,根据以前的研究(35),暗示了在ad患者的cns中hthnr的活化水平降低。因此,作为进一步的推论,提出了在ad患者的cns中作为hthnr的核心激动剂的clsp水平有可能降低。然而,根据本发明人不久之前的研究(14),否定了clsp水平本身在ad患者的cns中降低的可能性。
5.关于人体肽和clsp、以及它们的作用/效果,除上述以数字引用的参考文献(references)以外,在专利文献1中也有详细记载。
6.另一方面,脂连蛋白是来自脂肪组织的肽激素,其与脂连蛋白r1和脂连蛋白r2等受体结合以活化amp激酶介导的细胞内信号,从而显示增加胰岛素敏感性、胰岛素非依赖性的葡萄糖摄入、和分解脂肪酸等各种代谢作用。结果认为:该激素发挥抑制ii型糖尿病、肥胖、动脉粥样硬化、非酒精性脂肪性肝病、以及代谢综合征和相关的代谢异常的作用。
7.如下所述,脂连蛋白的缺乏或脂连蛋白信号传递的异常调节与ad的发病相关的间
接性证据在多个研究中以初步数据的形式展示(31)。血清脂连蛋白水平的上升(29、30)有可能是ad的独立的危险因子(32)。反之,通过研究显示:在血清脂连蛋白浓度低的ii型糖尿病患者中出现ad样病情(33)。在ad患者的csf中脂连蛋白水平降低,与aβ水平的增加逆相关(30)。脂连蛋白敲除小鼠显示ad样症状和病理所见(34)。
8.现有技术文献专利文献专利文献1:日本专利第5939528号说明书。
技术实现要素:
9.发明所要解决的课题研究显示:除hthnr以外,clsp还与多种蛋白结合(15),但它们的结合如何影响clsp功能尚未明确。
10.本发明的第一课题在于:研究这些clsp结合因子和本发明中新发现的clsp结合因子调节clsp活性的可能性,并对调节的蛋白进行其详细的机制分析。
11.第二课题在于:使用来自ad患者的样品确认在ad的中枢神经系统中clsp活性降低,同时研究这些clsp结合因子的异常有助于ad发病的可能性。
12.而且,第三课题在于提供:不受阻碍剂对clsp活性的阻碍或抑制作用的clsp衍生物、clsp和该clsp衍生物所具有的clsp活性的增强或保护剂、clsp或该clsp衍生物与增强或保护剂的融合蛋白、以及包含它们作为有效成分的用于抑制与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的药物组合物等。
13.用于解决课题的手段为了解决上述课题,本发明人深入研究的结果,在该技术领域中初次得到以下的见解,从而完成了本发明。
14.首先,发现clsp活性被载脂蛋白e (apolipoproteine:apoe;本实验中使用的apoe3和apoe4是有一个氨基酸不同的同族蛋白,生物化学性质基本相同)、14-3-3蛋白和钙网蛋白(calreticulin)等clsp阻碍物质(剂)抑制(图2和图3)。一系列的数据证实:这些clsp阻碍物质在与培养基中的clsp浓度同等~5倍的浓度下显示完全的clsp抑制效果。已知在人中枢神经系统中存在浓度绝对高于clsp浓度的apoe。例如,推测人脑脊液(csf)中的apoe浓度为40-200nm (18、19),另一方面,推测clsp的浓度为3-6nm (14)。因此,若假定在体内的cns中clsp活性由仅由clsp及其阻碍物质构成的单纯体系规定,则认为通过这样的高浓度的内源性apoe使clsp的活性完全无效(实际上在正常生物体内,如后所述存在clsp保护物质以维持clsp活性(图5、图6、图7)。因此,作为治疗手段,为了通过使野生型clsp增加的方法体现在ad的中枢神经系统中降低的clsp活性(图10、11、表1、2、3、4),必须使cns中的clsp浓度至少增加至40-200nm以上以克服clsp阻碍物质的阻碍。然而,由于clsp无法有效地透过血脑屏障而进入中枢神经系统(5、14),所以这难以通过从外周路径给予野生型clsp来达成。例如,在小鼠中,在腹腔内注射5nmol野生型clsp的1小时后(通常预测达到最大浓度) csf和血清中的clsp浓度分别达到5nm和500nm (5)。因此,根据单纯的计算,为了在csf中上升至40-200nm以上的浓度,需要至少给予约10倍以上的野生型clsp。然而,上述实验中给予的5nmol对小鼠而言已经是非常多的量,现实中难以增加其以上给药量。即,通
过从外周注射野生型clsp而在cns中出现clsp活性几乎是不可能的。因此,为了通过外周给予clsp而在cns中出现clsp活性,必须进行如下的变更或设计:使clsp更有效地透过血脑屏障、和/或使clsp从clsp阻碍物质的阻碍效果中解放出来。
15.本发明人发现:apoe4经由clsp的c末端区(氨基酸62-146)与clsp结合(图9和补充图s4)。该见解显示:clsp的n末端区(氨基酸1-61:简称“clsp1-61”)不与apoe结合,而且不受apoe介导的抑制。重要的是,本发明人进一步证实:clsp1-61具有与野生型clsp同等的活性,抑制v642i-app诱导性神经细胞死亡(图l1)。实际上,完全阻碍v642i-app诱导性神经元死亡的、在大肠杆菌中产生的clsp1-61和野生型clsp的最小必需浓度同为0.5nm (图l1和图2)。
16.不出所料,由clsp1-61介导的v642i-app诱导性神经细胞死亡的抑制不仅不受apoe3的阻碍,也不受14-3-3σ蛋白或钙网蛋白这样的其他clsp阻碍剂的阻碍(图l2)。由以上证实:clsp1-61完全从clsp阻碍物质的抑制中解放出来,而且活性与野生型几乎同等,因此其是在体内以远低于野生型clsp的浓度显示clsp活性的clsp衍生物。
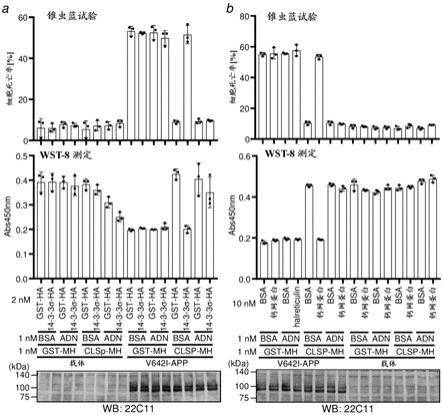
17.[seq id no: 1 clsp (1-146)]mageltpeeeaqykkafsavdtdgngtinaqelgaalkatgknlseaqlrklisevdsdgdgeisfqefltaakkaragledlqvafrafdqdgdghitvdelrramaglgqplpqeeldamireadvdqdgrvnyeefarmlaqe (根据遗传性多态性,第58位的s有时是g,但活性相同)进一步发现:脂连蛋白通过与clsp的her (内源性人体肽同源区)结合(图1和图9、图s4),增强clsp活性(活性增强因子;图7),并且保护(保持) clsp活性免受所有种类的clsp阻碍物质的阻碍(活性保护因子;图5、图6)。实际上,即使存在高达50nm的浓度绝对高的clsp阻碍物质,但如果存在0.2-0.25nm浓度的脂连蛋白,则ad相关细胞死亡也完全被1nm的clsp抑制(图5和图7)。该结果显示:在存在浓度绝对高于clsp的clsp阻碍物质的cns中,脂连蛋白是保持clsp的活性的clsp活性保护因子。
[0018]
进一步发现:通过使用临床样品进行实验,在ad患者中csf中的脂连蛋白的水平降低至0.3nm (图10、表1和2)。该结果与以前的研究结果一致(30)。还发现在ad患者中神经元内clsp信号强度降低(图11、表3和4)。若将这些结果一并考虑在内,则暗示在ad患者中因某种原因导致cns中的脂连蛋白水平降低,其结果是clsp活性降低、神经元对ad相关毒性敏感(即产生神经毒性)。
[0019]
本发明人还发现:脂连蛋白(adn)的胶原同源区(adncol:相当于and中的第45~104位的氨基酸序列)单独与clsp结合(图s4),就足以显示clsp增强/保护活性(图l3和图l4)。重要的是,adncol的clsp增强/保护活性仅稍弱于野生型脂连蛋白的该活性的这个事实。实际上,用于赋予完全的clsp增强/保护活性的野生型脂连蛋白的最小浓度为0.2-0.25nm,而adncol的该最小浓度为0.5nm。另外,已知位于脂连蛋白的c末端的球形结构域对由普通的脂连蛋白受体adipor1和2介导的葡萄糖降低效果等脂连蛋白的代谢活性的调节是必须的(42)。因此,缺失了球形结构域的adncol缺乏脂连蛋白的这些代谢效果。即,缺失了球形结构域的adncol虽然与野生型adn同样具有完全的clsp活性增强/保护作用,但不同于野生型and,其无法与普通的脂连蛋白受体结合,其结果,认为没有显示所谓的代谢调节活性(可成为副作用的活性)。另一方面,在以前发表的研究(33、34)中,推测脂连蛋白的抗ad活性是由通过普通的脂连蛋白受体adipor1和2的结合而引起的代谢调节活性介导的。
[0020]
由上述内容预测:相对于野生型脂连蛋白,作为clsp增强/保护剂的adncol具有4个优点。第一,考虑到体内组织(cns或外周组织)中的普通脂连蛋白受体adipor1和2 (普通型脂连蛋白受体)的富裕度,推测有相当比例的野生型脂连蛋白被消耗于与普通型脂连蛋白受体形成复合体,相对于此adncol却不是这样。第二,暗示ad中的csf脂连蛋白水平的降低可能是源于因与神经元内的过度磷酸化tau形成不溶性复合体而被消耗(30),但推测该过程是通过野生型脂连蛋白与普通型脂连蛋白受体结合且被神经元摄入而引起的。由于adncol不与普通型脂连蛋白受体结合,因此很可能不会与神经元中的过度磷酸化tau形成复合体。以上两点暗示:在体内显示clsp增强/保护活性所需的adncol量要较野生型脂连蛋白少。第三,大量的野生型脂连蛋白有可能通过与普通型脂连蛋白受体结合且活化各种代谢路径而引起副作用,但adncol不与普通受体结合,因此推测没有这样的副作用。第四,与野生型脂连蛋白的氨基酸长度(244个氨基酸:seq id no: 3)相比,adncol的氨基酸长度(60个氨基酸:seq id no: 2)相对较短,因此容易进行工业生产。根据上述的adncol的全部优点,adncol作为抗ad药也比野生型脂连蛋白优异。
[0021]
另外,本发明人发现:clsp或clsp衍生物与增强或保护剂的融合蛋白(杂交肽)对v642i-app诱导性神经细胞死亡具有较clsp1-61和野生型clsp强的保护活性(图l5)。即,用于完全抑制v642i-app诱导神经元细胞死亡的由clsp1-61和adncol构成的杂交肽(命名为“clspcol”)和由野生型clsp和adncol构成的杂交肽(命名为“wt-clspcol”)的最小浓度为0.1nm,clsp1-61和野生型clsp的最小浓度为0.5nm (图l5)。另外,clspcol和wt-clspcol不被clsp阻碍剂抑制,即使被抑制其程度也是轻度的(图x1和x2)。
[0022]
进一步发现:clspcol比wt-clspcol更有效地透过血脑屏障而转移到cns中(图l6和表1)。即,在小鼠中腹腔内注射10nmol的clspcol的1小时后,clspcol的浓度在含间质液(isf)的脑匀浆中为72nm、在血清中为320nm (图l6和表l1)。
[0023]
[adncol:seq id no: 2]ghpghngapgrdgrdgtpgekgekgdpgligpkgdigetgvpgaegprgfpgiqgrkgep[adn:seq id no: 3]mlllgavllllalpghdqetttqgpgvllplpkgactgwmagipghpghngapgrdgrdgtpgekgekgdpgligpkgdigetgvpgaegprgfpgiqgrkgepgegayvyrsafsvgletyvtipnmpirftkifynqqnhydgstgkfhcnipglyyfayhitvymkdvkvslfkkdkamlftydqyqennvdqasgsvllhlevgdqvwlqvygegernglyadndndstftgfllyhdtn即,本发明涉及以下方案。
[0024]
[方案1]钙调素样皮肤蛋白(calmodulin-like skin protein:clsp)衍生物(突变体),所述衍生物的特征在于:包含内源性人体肽同源区(ehr),该her是抑制与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的活性(clsp活性)中心,不包含该clsp活性阻碍剂所结合的区域。
[0025]
[方案2]方案1所述的衍生物,其中,her由氨基酸序列(i)构成:tgknlseaqlrklisevds (或g) dgd (氨基酸单字母表示) (i)。
[0026]
[方案3]方案1或2所述的衍生物,其中,阻碍剂所结合的区域是clsp (seq id no: 1)的c末端区的氨基酸序列(氨基酸62~146)。
[0027]
[方案4]方案1~3中任一项所述的衍生物,其是由以下的氨基酸序列构成的多肽:(1) clsp的n末端区的氨基酸序列(氨基酸1~61);(2) 上述(1)的氨基酸序列中,在该氨基酸序列所含的ehr以外的氨基酸序列中有一个或多个(例如,2~5个左右)氨基酸被缺失、取代或插入而得的氨基酸序列;或者(3) 上述(1)的氨基酸序列中,相对于该氨基酸序列所含的ehr以外的氨基酸序列具有90%以上、优选95%以上、进一步优选98%以上的同一性的氨基酸序列。
[0028]
[方案5]方案1~4中任一项所述的衍生物,该衍生物不受阻碍剂对clsp活性的阻碍或抑制作用。
[0029]
[方案6]方案1~5中任一项所述的衍生物,其中,阻碍剂选自载脂蛋白e、14-3-3蛋白和钙网蛋白。
[0030]
[方案7]多肽,其由以下的氨基酸序列构成:(1) seq id no: 2所示的氨基酸序列(adncol);(2) 包含上述(1)的氨基酸序列(adncol)的氨基酸序列;(3) seq id no: 3所示的脂连蛋白的氨基酸序列中,在该氨基酸序列所含的adncol以外的氨基酸序列中有一个或多个氨基酸被缺失、取代或插入而得的氨基酸序列;或者(4) 在seq id no: 3所示的脂连蛋白的氨基酸序列中,相对于该氨基酸序列所含的adncol以外的氨基酸序列具有90%以上的同一性的氨基酸序列。
[0031]
[方案8]clsp或方案1所述的clsp衍生物所具有的clsp活性的增强或保护剂,其由方案7所述的多肽构成。
[0032]
[方案9]方案8所述的增强或保护剂,其特征在于:保护该clsp免受阻碍剂对clsp活性的阻碍或抑制作用,或者,使阻碍剂的该作用失效。
[0033]
[方案10]方案8或9所述的增强或保护剂,其中,上述多肽为脂连蛋白。
[0034]
[方案11]方案8~10中任一项所述的增强或保护剂,其中,阻碍剂选自载脂蛋白e、14-3-3蛋白和钙网蛋白。
[0035]
[方案12]融合蛋白,其包含clsp或方案1所述的clsp衍生物和方案7所述的多肽。
[0036]
[方案13]
方案12所述的融合蛋白,其由clsp的n末端区的氨基酸序列(氨基酸1~61)和adncol构成。
[0037]
[方案14]方案12或13所述的融合蛋白,其不受阻碍剂对clsp活性的阻碍或抑制作用。
[0038]
[方案15]药物组合物,其用于抑制与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡,包含方案1~6中任一项所述的clsp衍生物、方案7所述的多肽、方案8~11中任一项所述的增强或保护剂、或方案12~14中任一项所述的融合蛋白作为有效成分。
[0039]
[方案16]方案15所述的药物组合物,其用于预防或治疗伴有与阿尔茨海默病相关的记忆损伤或神经变性的疾病。
[0040]
[方案17]治疗伴有神经细胞的细胞功能障碍或神经细胞死亡的疾病、或伴有记忆损伤或神经变性的病症的方法,该方法包括:对患有或疑似患有该疾病或病症的个体给予方案15或16所述的药物组合物的阶段。
[0041]
[方案18]方案17所述的方法,其中,疾病或病症为阿尔茨海默病。
[0042]
[方案19]检测方案1~6中任一项所述的clsp衍生物、方案7所述的多肽、或方案8~11中任一项所述的增强或保护剂、或方案12~14中任一项所述的融合蛋白(将以上统称为“本发明多肽”)对与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的抑制活性的方法,该方法包括:工序(a),在clsp的阻碍剂的存在/不存在下、和本发明多肽的存在/不存在下,诱导神经细胞的功能障碍或神经细胞死亡;工序(b),检测神经细胞的功能障碍或神经细胞死亡;以及工序(c),比较在本发明多肽的存在/不存在下的神经细胞的功能障碍或神经细胞死亡。
[0043]
[方案20]筛选调节方案1~6中任一项所述的clsp衍生物、方案7所述的多肽、或方案8~11中任一项所述的增强或保护剂、或方案12~14中任一项所述的融合蛋白(将以上统称为“本发明多肽”)或clsp对与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的抑制活性的物质的方法,该方法包括:工序(a),在本发明多肽或clsp的存在下、在受试物质的有无下诱导神经细胞的功能障碍或神经细胞死亡;工序(b),检测神经细胞的功能障碍或神经细胞死亡;以及工序(c),选择调节本发明多肽或clsp对神经细胞的功能障碍或神经细胞死亡的抑制活性的物质。
[0044]
发明效果本发明的clsp衍生物包含内源性人体肽同源区(ehr),该ehr是抑制与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的活性(clsp活性)中心,不包含像apoe、或14-3-3σ蛋白或钙网蛋白这样的clsp活性阻碍剂所结合的区域。
[0045]
其结果,clsp衍生物具有与野生型clsp相同程度的clsp活性,并且,实质上(显著)
不受该阻碍剂对clsp活性的阻碍或抑制作用。由以上显示:这些多肽被完全从clsp阻碍剂的阻碍/抑制中解放出来,在体内以远低于野生型clsp的浓度显示clsp活性。
[0046]
另一方面,由seq id no: 2所示的氨基酸序列构成且作为脂连蛋白的胶原同源区的多肽、和包含seq id no: 2所示的氨基酸序列的多肽、例如三聚体等多聚体脂连蛋白与位于clsp和本发明的clsp衍生物的clsp1-61内的ehr结合,具有增强它们所具有的clsp活性的作用/效果。
[0047]
而且,上述多肽具有以下的作用/效果:保护clsp免受载脂蛋白e等阻碍剂对clsp活性的阻碍或抑制,或者使该阻碍剂的阻碍或抑制作用失效。因此,上述多肽可用作与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的抑制活性的增强或保护剂。
[0048]
另外,本发明的融合蛋白具有较由clsp或clsp的一部分构成的衍生物强的抗ad活性。另外,融合蛋白不受clsp阻碍剂的阻碍,或者即使受到阻碍也是极轻度的水平。进一步预测:该肽缺乏来自脂连蛋白的代谢相关活性,而且不会因与标准的脂连蛋白受体形成复合体而被消耗。除这些优点之外,作为融合蛋白之一的clspcol还具有其血脑屏障移动非常好的特征,所以可成为可外周给药的理想的抗ad药的可能性高。
附图说明
[0049]
[图1] 《载脂蛋白e3、e4和脂连蛋白与clsp结合》 通过转染使c末端用ha标记的载脂蛋白e3、e4、脂连蛋白和膜联蛋白ii在f11神经杂交细胞中过度表达。在转染的24小时后,回收f11细胞,调制细胞裂解物。相对于300μg的裂解物加入另外调整的结合有适量的gst-mychis或clsp-mychis的sepharose (琼脂糖凝胶) 4b,在4℃下培养一夜,彻底洗涤,然后进行pull-down沉降(沉淀)。将由细胞裂解物或与gst-mychis (gst-mh)和clsp-mychis (clsp-mh)结合的sepharose 4b珠粒构成的input、以及细胞裂解物的pull-down沉降物进行sds-page展开后,使用ha (血凝素a)和myc抗体进行免疫印迹分析。
[0050]
[图2] 《载脂蛋白e3和e4抑制clsp活性》 (a) 对于sh-sy5y细胞,转染pcdna3.1/mychis载体(载体)或pcdna3.2/mychis-v642i-app (v642i-app)。然后,在含有所示浓度的clsp-mychis的dmem/f12-10�s中进行培养。转染的24小时后,将培养基交换为包含含有相同浓度的clsp-mychis的n2补充剂的dmem/f12。在转染开始的48小时后,进行使用wst-8细胞死亡测定试剂盒的细胞存活测定、或钙黄绿素am染色、和锥虫蓝排除细胞死亡测定。另外,使用app抗体22c11对细胞裂解物进行免疫印迹分析。(b、c) 对于sh-sy5y细胞,转染pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)。然后,在包含/不含所示浓度的bsa、载脂蛋白e3 (b)或e4 (c)且含有1nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含/不含相同浓度的bsa、载脂蛋白e3 (b)或e4 (c)且包含含有1nm的gst-mychis或gst-mychis的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡测定。另外,使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0051]
[图3] 《14-3-3家族蛋白和分泌型钙网蛋白抑制clsp活性》 (a-e) 对于sh-sy5y细胞,转染pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)。然后,在包含/不含所示浓度的bsa和14-3-3同种型且含有10nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。在转染的24小时后,将培养基交换为包含/不含相同浓度
的bsa或14-3-3同种型且包含含有10nm的gst-mychis或clsp-mychis的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡测定。(f) 使用空的pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染sh-sy5y细胞。然后,在包含/不含10nm的bsa、钙网蛋白、膜联蛋白ii或膜联蛋白v且包含10nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。在转染的24小时后,将培养基交换为包含/不含10nm的bsa、钙网蛋白、膜联蛋白ii或膜联蛋白v且包含含有相同浓度的gst-mychis或clsp-mychis的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0052]
[图4] 《脂连蛋白保护clsp活性免受载脂蛋白e3的阻碍》 (a-c) 使用空的pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染sh-sy5y细胞。然后,在包含/不含10nm的脂连蛋白(a)、膜联蛋白ii(b)或膜联蛋白v(c)且包含1nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后实施锥虫蓝排除细胞死亡测定。另外,使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0053]
[图5] 《脂连蛋白保护clsp活性免受载脂蛋白e4的阻碍》 (a) 对于sh-sy5y细胞,转染pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app/mychis-v642i-app (v642i-app)。然后,在包含/不含所示浓度的脂连蛋白、包含/不含10nm的载脂蛋白e4且包含1nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后回收细胞,实施wst-8和锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。(b) 将pcdna3.1/mychis载体(载体)或pcdna3.1/ mychis-v642i
‑ꢀ
app (v642i-app)转染到sh-sy5y细胞中。然后,在包含/不含1nm的脂连蛋白、包含/不含浓度阶段性地增加的载脂蛋白e4、且含有1nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后回收细胞,实施wst-8和锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0054]
[图6] 《脂连蛋白保护clsp免受14-3-3σ和钙网蛋白的阻碍》 (a、b) 使用pcdna3载体(载体)或pcdna3-v642i-app (v642i-app)对sh-sy5y细胞进行转染。然后,在包含/不含2nm的14-3-3σ(a)或10nm的钙网蛋白(b)、包含/不含1nm的脂连蛋白且含有1nm的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后回收细胞,实施锥虫蓝排除细胞死亡和wst-8测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0055]
[图7] 《脂连蛋白增强clsp活性》 (a) 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,在于含有所示浓度的gst-mychis或clsp-mychis的dmem/ f12-10�s中包含/不含200pm的脂连蛋白的培养液中培养细胞。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。(b) 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i
‑ꢀ
app (v642i-app)转染到sh-sy5y细胞中。接下来,在包含/不含所示浓度的脂连蛋白、且包含所示浓度的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交
换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡和wst-8测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0056]
[图8] 《脂连蛋白与clsp的结合的解离常数和载脂蛋白e4与clsp的结合的解离常数近似》 (a) 脂连蛋白的存在仅限于轻微抑制载脂蛋白e4与clsp之间的结合。将包含结合有clsp-mychis的sepharose 4b的pbs与脂连蛋白、膜联蛋白ii、重组载脂蛋白e3或e4中的任一种或两种进行混合,在4℃下培养一夜后充分洗涤。测定中的各重组蛋白的最终推测浓度为1nm。然后,进行pull-down,将所生成的沉淀物、以及结合有clsp-mychis的sepharose 4b珠粒和各重组蛋白作为input进行sds-page展开,然后通过银染色将其可视化。(b) 进行斯卡查德分析以测定解离常数。将用浓度20pm的重组载脂蛋白e4或脂连蛋白包被的96孔板的各孔用浓度阶段性地增加的clsp-hibit填满,在室温下培养2小时后,使用wallac arvo
tm x5 (perkin elmer)进行化学发光测定,推测hibit活性。该实验以n=2进行实施,将2个孔的平均数据(平均)用于进一步的分析。从各平均clsp-hibit活性(平均)中扣除零浓度下的平均clsp-hibit活性(背景),得到了与脂连蛋白或载脂蛋白e4结合的实际的clsp-hibit活性(del/mean)。然后,参照由clsp-hibit浓度和对应的化学发光强度(即clsp-hibit活性)构成的标准的剂量反应曲线,推测与载脂蛋白e4或脂连蛋白结合的clsp-hibit的浓度(用《b》表示)。然后,计算游离clsp-hibit浓度(非结合浓度) (以《f》的形式表示)和b/f。解离常数通过使用prism7软件的斯卡查德分析进行计算。
[0057]
[图9] 《载脂蛋白e4和脂连蛋白与clsp的不同位点结合》 (a) 显示clsp的缺失突变体的简图。(b) 通过转染使c末端用flag标记的载脂蛋白e4 (apoe4)和脂连蛋白(adn)在f11神经杂交细胞中过度表达。在转染的24小时后回收f11细胞,调制细胞裂解物。在使用flag抗体的apoe4-flag和adn-flag的免疫沉降中使用300μg的细胞裂解物。另外,使重组clsp-mychis (fl-mh)或c末端带有mychis标签的clsp缺失突变体在细菌中产生并纯化。然后,将这些免疫沉降物和重组蛋白作为input使用myc抗体和flag抗体进行sds-page展开,进行免疫印迹分析。(c) 将(b)中制作的apoe4-flag和adn-flag免疫沉降物与纯化重组clsp-mychis (fl-mh)或c末端mychis标签clsp缺失突变体进行混合,在4℃下培养一夜,然后彻底洗涤。然后,将pull-down沉淀物使用myc抗体和flag抗体进行sds-page展开,进行免疫印迹分析。
[0058]
[图10] 《在ad患者的csf中脂连蛋白降低》 (a) 使用脂连蛋白elisa系统测定表1所示的由ad患者和非ad对照得到的csf中的脂连蛋白浓度。标准的剂量-响应曲线通过测定重组脂连蛋白的阶段性增加的浓度而制作。(b) 以ad患者和非ad对照中的各csf脂连蛋白浓度为点作图(ad病例n=14、非ad病例n=20)。还显示了脂连蛋白浓度的平均
±
sem (ad、0.31
±
0.13nm;非ad、0.96
±
0.19nm;非配对t检验、p=0.0065)。(c) 以81~88岁的ad患者和非ad对照中的各csf脂连蛋白浓度为点作图(ad病例n=6、非ad病例n=5)。还显示了脂连蛋白浓度的平均
±
sem (ad、0.30
±
0.07nm;非ad、1.41
±
0.16nm;非配对t检验、p 《0.0001)。
[0059]
[图11] 《ad皮质的神经元内sh3bp5水平降低》 (a) 将来自2名ad患者(65岁男性;79岁女性)和als患者(66岁男性;79岁男性)的颞叶或枕叶的外锥体层用抗sh3bp5抗体进行免疫染色。免疫检测按照酪酰胺红法来进行。比例尺为200mm。(b) 细胞的免疫荧光强度的定量化例子。用标记包围细胞区和细胞周围的非细胞区,测定细胞区的平均免疫荧光强度
(x)和细胞周围的非细胞区的平均免疫荧光强度(y)。然后,通过(x-y)计算神经元中的相对平均免疫荧光强度,用神经元面积乘以x-y值,计算一个神经元中的sh3bp5表达水平。(c) 如表2所示,将来自ad患者和肌萎缩性侧索硬化症(als)患者的颞叶或枕叶的外锥体层的切片(包括(a)所示的切片)用抗sh3bp5抗体进行免疫染色。免疫检测按照酪酰胺红法来进行。如“材料和方法”中详细所述,利用image j 1.37v测定免疫荧光强度。以ad患者和als患者中的各相对强度为点作图(ad病例n=7、als病例n=6)。还显示了免疫荧光强度的平均
±
sem (ad、46564
±
7737任意单位;als、79225
±
10305任意单位;非配对t检验、p = 0.0256)。(d、e) 如表3所示,利用sh3bp5 elisa测定由ad患者和非ad的颞叶皮质得到的20μl裂解物中的sh3bp5浓度。通过测定浓度阶段性增加的重组sh3bp5,制作标准的剂量反应曲线(d)。将ad患者和非ad中的sh3bp5的相对浓度作图(各组n=10) (e)。还显示了相对的sh3bp5水平的平均值
±
sd (ad、103.9
±
9.0任意单位;正常、159.4
±
16.5任意单位;非配对t检验、p=0.0084)。
[0060]
[图12] 图s1 (补充图1) 《脂连蛋白本身没有阻碍v642i-app诱导性神经细胞死亡,没有阻碍由clsp介导的v642i-app诱导性神经细胞死亡的减少》 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i
‑ꢀ
app (v642i-app)转染到sh-sy5y细胞中。接下来,在包含/不含浓度阶段性增加的脂连蛋白、且含有gst-mh或clsp-mh的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含/不含浓度阶段性增加的脂连蛋白、且含有包含gst-mh或clsp-mh的n2补充剂的dmem/f12。从转染开始起48小时后回收细胞,采用wst-8细胞死亡测定试剂盒(同仁堂、熊本、日本)或钙黄绿素am染色(同仁堂)和锥虫蓝排除细胞死亡测定实施细胞存活测定。另外,使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0061]
[图13] 图s2 (补充图2) 《人csf中的14-3-3σ水平为检测限以下》 (a) 使用14-3-3σ elisa系统测定由8名非ad患者(csf#1-8)得到的20μl csf中的14-3-3σ浓度。实验进行2次。浓度阶段性增加的标准14-3-3σ (浓度;0.195~6.25nm)和8名非ad患者的csf的原始测定数见abs450列。接下来,计算2个数值的平均,见平均abs450列。使用pbs作为阴性对照。通过从各平均值(平均数)中扣除pbs值,得到了del abs 450nm值。(b) 标准的剂量-响应曲线通过测定重组14-3-3σ的阶段性增加的浓度而制作。由此推测:利用该elisa可检测的最低限为0.4nm。(a)中的del ab 450nm的各数据的csf 14-3-3σ浓度为检测限以下。
[0062]
[图14] 图s3 (补充图3) 《三聚体脂连蛋白具有与野生型脂连蛋白相媲美的clsp活化效果》 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,在包含/不含1nm三聚体或野生型(单)脂连蛋白、且含有所示浓度的gst-mychis或clsp-mychis的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施wst-8测定和锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。“***”p 《0.001;“n.s.”不显著。
[0063]
[图15] 图s4 (补充图4) 《clsp与apoe4或脂连蛋白的结合的详细分析》 (a、b) 《载脂蛋白e4与clsp的c末端区结合》 clsp的缺失突变体的简图见(a)。通过转染使c末端用flag标记的载脂蛋白e4 (apoe4)和脂连蛋白(adn)在f11神经杂交细胞中过度表达。在转染的24小时后将f11细胞调制成细胞裂解物。在使用flag抗体的apoe4-flag和adn-flag的免疫沉降中,使用300μg的细胞裂解物。使重组clsp-mychis (fl-mh)或c末端带有mychis标签
的clsp缺失突变体在细菌中产生,然后进行纯化。接下来,使用myc和flag抗体对这些免疫沉降物和重组蛋白进行sds-page和免疫印迹分析(input)。将apoe4-flag和adn-flag免疫沉降物与重组clsp-mychis (fl-mh)或c末端带有mychis标签的clsp缺失突变体进行混合,在4℃下培养一夜,然后彻底洗涤。然后,使用myc抗体和flag抗体将pull-down沉淀物进行sds-page展开,进行免疫印迹分析。(c) 《clsp与脂连蛋白的胶原同源区结合》 使n末端用6
×
his和g (hisg)标记的脂连蛋白的胶原同源区(adncol)在细菌中产生。另外,通过转染使clsp-flag在f11神经杂交细胞中过度表达。使用hisg-adncol以及flag抗体,将免疫沉降的纯化重组flag-clsp和对照(载体)进行sds-page展开,使用flag和hisg抗体进行免疫印迹分析(input;左图)。接下来,混合已纯化的重组hisg-andcol和免疫沉降的clsp-flag或对照(载体),然后在4℃下培养一夜,之后彻底洗涤。然后,使用flag抗体和hisg抗体将pull-down沉淀物进行sds-page展开,进行免疫印迹分析(co-ip;右图)。
[0064]
[图16] 图s5 (补充图5) 《年龄与csf脂连蛋白浓度之间无关》 针对表1和表s1的全部对象将脂连蛋白水平和年龄的原始数据作图(x轴:年龄;y轴:csf脂连蛋白浓度)。相关系数为0.0055。
[0065]
[图17] 图s6 (补充图6) 《神经元中的sh3bp5水平不受高龄的影响》 将图11c所示的关于ad患者或als患者的sh3bp5数据根据年龄分成2组,进行比较。一组由70岁以下的人构成,另一组由71岁以上的人构成。2组的sh3bp5的平均
±
sem相对强度为57439
±
14465任意单位和65237
±
7976任意单位(非配对t检验、p=0.6328、t=0.49、r二乗=0.021、自由度=11、f检验的p值=0.24)。
[0066]
[图18] 图l1 《完全抑制v642i-app诱导神经细胞死亡的clsp1-61的最小浓度为500pm》 (a、b) 通过pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)对sh-sy5y细胞进行转染。然后,在包含所示浓度的gst-mychis或clsp (1-61)-mychis的dmem/f12-10�s中培养细胞。在转染的24小时后,将培养基交换为包含含有相同浓度的gst-mychis或clsp (1-61)-mychis的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡率、wst8和钙黄绿素测定。另外,使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0067]
[图19] 图l2 《clsp阻碍物质不阻碍clsp1-61的v642i-app诱导神经细胞死亡抑制效果》 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,将细胞在同时包含10nm的bsa、apoe3、14-3-3σ或钙网蛋白和1nm的gst-mychis或clsp (1-61)-mychis的dmem/f12-10�s中进行培养,在转染的24小时后,将培养基交换为包含同时含有相同浓度的bsa、apoe3、14-3-3σ或钙网蛋白和gst-mychis或clsp(1-61)-mychis的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡率、wst8和钙黄绿素测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0068]
[图20] 图l3 《脂连蛋白的胶原同源区增强clsp活性》 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,将细胞在同时含有1nm的bsa、脂连蛋白(fl)或脂连蛋白的胶原同源区(col)和1nm或50pm的gst-mychis或clsp-mychis的dmem/f12-10�s中进行培养。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem /f12。从转染开始起48小时后采集细胞,实施
锥虫蓝排除细胞死亡率、wst8和钙黄绿素测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0069]
[图21] 图l4 《完全活化50pm的clsp的脂连蛋白的胶原同源区的最小浓度为500pm》 将pcdna3.1/mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,将细胞在包含500μm的bsa、250μm的脂连蛋白(fl)或所示浓度的脂连蛋白的胶原同源区(col)、且包含50pm的gst-mychis或clsp-mychis的dmem/f12-10�s中进行培养。转染的24小时后,将培养基交换为包含含有相同组合的蛋白的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡率、wst8和钙黄绿素测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0070]
[图22] 图l5 《clspcol具有强效的ad保护活性》 将pcdna3.1mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,在含有1nm的gst-mychis、clsp1-61-mychis、clsp-mychis或所示浓度的clspcol或wt-clspcol的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同浓度的试剂的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0071]
[图23] 图l6 《clspcol有效地透过血脑屏障》 (a) 如表l1所示,通过针对浓度阶段性增加的wt-clspcol和clspcol在450nm下测定吸光度,模拟标准剂量反应曲线。(b) 在腹腔内注射10nmol的gst-mychisg、clspcol和wt-clspcol的1小时后,由小鼠采集脑和血清以进行elisa。如表l1所示,利用elisa测定包含间质液(isf)的脑裂解物和血清中的clspcol和wt-clspcol的浓度。(c) 计算isf与血清浓度之比,进行展示。
[0072]
[图24] 图x1 《clspcol虽然不被apoe3和14-3-3σ阻碍,但被钙网蛋白轻度阻碍》 将pcdna3.1mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,在含有100pm的gst-mychis或clspcol和1nm的apoe3、14-3-3σ、或钙网蛋白的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同浓度的试剂的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
[0073]
[图25] 图x2 《clspcol开始被10倍以上的高浓度的钙网蛋白阻碍》 将pcdna3.1mychis载体(载体)或pcdna3.1/mychis-v642i-app (v642i-app)转染到sh-sy5y细胞中。然后,在含有gst-mychis (1nm)、clsp1-61-mychis (1nm)、clspcol (100pm)或wt-clspcol (100pm)和所示浓度的钙网蛋白或bsa的dmem/f12-10�s中培养细胞。转染的24小时后,将培养基交换为包含含有相同浓度的试剂的n2补充剂的dmem/f12。从转染开始起48小时后采集细胞,实施锥虫蓝排除细胞死亡测定。使用app抗体22c11对细胞裂解物进行免疫印迹分析。
具体实施方式
[0074]
[clsp衍生物]由钙调素样皮肤蛋白(calmodulin-like skin protein:clsp) (氨基酸序列1)所含的22个氨基酸(氨基酸40~61)构成的氨基酸序列(i):tgknlseaqlrklisevds (或g) dgd (氨基酸单字母表示) (i)被称为内源性人体
肽同源区(endogenous humanin-homogenous region:ehr)或内源性人体肽样结构域((endogenous humanin-like domain:ehd),在clsp介导的神经细胞死亡抑制中发挥核心作用(专利文献1)。
[0075]
本发明的clsp衍生物的特征在于:包含作为抑制与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的活性(clsp活性或clsp抑制活性)中心的内源性人体肽同源区(ehr),不包含该活性的阻碍剂或阻碍物质(clsp阻碍剂)所结合的区域。作为该阻碍剂所结合的区域,例如可列举:clsp (seq id no: 1)的c末端区的氨基酸序列(氨基酸62~146)。
[0076]
本发明中,“抑制与阿尔茨海默病相关的神经细胞的功能障碍或细胞死亡的活性”是指抑制或拮抗神经细胞中的功能障碍或细胞死亡的至少1者,而不依赖于其原因或因果关系。神经细胞死亡的抑制即使不是完全的抑制,只要显著地抑制即可。神经细胞死亡的抑制活性可按照以下的实施例中记载的方法或另外记载的方法(例如参照国际公开号 wo00/14204)进行检验。例如,clsp活性可采用各种神经细胞死亡测定进行测定,作为v642i-app诱导性神经细胞死亡的抑制活性。
[0077]
而且,阻碍剂与clsp的结合可利用如本说明书的实施例中记载的本领域技术人员已知的任意方法/手段(测定系统)进行测定。例如,可通过免疫印迹分析、pull-down分析、nano-glo hibit细胞外检测系统和elisa等进行测定。
[0078]
这里,作为ehr的具体例子,可列举:氨基酸序列(i):tgknlseaqlrklisevds (或g) dgd (氨基酸单字母表示) (i)、或由专利文献1的权利要求1所述的22个氨基酸构成的氨基酸序列。而且,作为阻碍剂所结合的区域的例子,可列举:clsp (seq id no: 1)的c末端区的氨基酸序列(氨基酸62~146)。
[0079]
因此,作为本发明的clsp衍生物的适合例子,可列举:由以下的氨基酸序列构成的多肽,(1) clsp的n末端区的氨基酸序列(氨基酸1~61);(2) 上述(1)的氨基酸序列中,在该氨基酸序列所含的ehr以外的氨基酸序列中有一个或多个(例如,2~5个左右)氨基酸被缺失、取代或插入而得的氨基酸序列;或者(3) 在上述(1)的氨基酸序列中,相对于该氨基酸序列所含的ehr以外的氨基酸序列具有90%以上、优选95%以上、进一步优选98%以上的同一性的氨基酸序列。
[0080]
本发明的clsp衍生物的特征在于:具有与野生型clsp相同程度的clsp活性,并且不包含阻碍剂所结合的区域,因此实质上(显著)不受阻碍剂对clsp活性的阻碍或抑制作用。尚需说明的是,本发明的clsp衍生物例如还包含缺失突变体等各种突变体和包含her的融合蛋白(杂交多肽)等,但不包含仅由her构成的多肽。
[0081]
另一方面,作为clsp阻碍剂,对其结构特征等没有特别限定,例如是在与培养基中的clsp浓度相同程度或5倍以上的浓度下显示对clsp活性的显著的阻碍(抑制)效果的物质,例如选自载脂蛋白e (apoe)、14-3-3蛋白和钙网蛋白。特别是显示apoe (apoe3和apoe4)的clsp活性抑制效果高。
[0082]
[脂连蛋白及其衍生物]而且,在本发明中判明:脂连蛋白(seq id no: 3)通过作为其胶原同源区(adncol)的多肽(seq id no: 2)与clsp和本发明的clsp衍生物的clsp1-61区内的ehr结
合,具有增强它们的clsp活性的作用/效果;而且,脂连蛋白和该多肽还具有以下作用:保护该clsp免受上述阻碍剂对clsp活性的阻碍或抑制作用、或使阻碍剂的该作用失效。
[0083]
因此,由以下的氨基酸序列构成的多肽(以下,也称为“脂连蛋白及其衍生物”)可用作clsp或本发明的clsp衍生物所具有的clsp活性的增强或保护剂:(1) seq id no: 2所示的氨基酸序列(adncol);(2) 包含上述(1)的氨基酸序列(adncol)的氨基酸序列、例如seq id no: 3所示的脂连蛋白;(3) seq id no: 3所示的脂连蛋白的氨基酸序列中,在该氨基酸序列所含的adncol以外的氨基酸序列中有一个或多个(例如,2~5个左右)氨基酸被缺失、取代或插入而得的氨基酸序列;或者(4) 在seq id no: 3所示的脂连蛋白的氨基酸序列中,相对于该氨基酸序列所含的adncol以外的氨基酸序列具有90%以上、优选95%以上、进一步优选98%以上的同一性的氨基酸序列。尚需说明的是,这些多肽例如可形成三聚体脂连蛋白这样的多聚体。
[0084]
尚需说明的是,“clsp”除含有本说明书中记载的seq id no: 1所示的多肽以外,还含有如专利文献1中记载的具有clsp活性的各种clsp的相关(类似)多肽。另外,关于这样的clsp活性,对于与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的“抑制”和“阻碍”同义。而且,关于本发明的增强或保护剂的活性,“保护”、“维持”和“保持”同义。
[0085]
[clsp衍生物等与脂连蛋白衍生物等的融合蛋白]而且,本发明还涉及融合蛋白(杂交多肽),其包含作为上述clsp衍生物的一例的clsp或clsp衍生物和脂连蛋白或脂连蛋白衍生物。该融合蛋白具有强效的clsp活性,不受clsp阻碍剂的抑制,即使受抑制也仅限于轻度。
[0086]
特别是作为该一适合例的、由clsp的n末端区的氨基酸序列(氨基酸1~61)和adncol构成的融合蛋白(clspcol)可有效地透过血脑屏障而转移到cns中。
[0087]
所涉及的融合蛋白只要不损及其所具有的规定的活性即可,可任意包含除构成上述各区(要素)的多肽以外的氨基酸序列。例如,为了提高蛋白三维结构的稳定性等,也可在各区之间插入由适当的氨基酸序列构成的接头序列。或者,例如以体内稳定性(血浆中的半衰期等) 等的提高等为目的,也可在c末端侧添加在已知的融合蛋白中发现的免疫球蛋白恒定区等本领域技术人员已知的任意的氨基酸序列。
[0088]
这样的追加/插入序列可由本领域技术人员根据技术常识边考虑抗原性等边适当地设计/调制。尚需说明的是,关于抗原性,由于clsp1-61和胶原同源区来自内源性人肽,所以推测它们的抗原性是有限的。
[0089]
而且,对融合蛋白中所含的各区的连接顺序(n末侧或c末侧)没有特别限定,本领域技术人员可适当地选择/调制。
[0090]
以下,将本发明的clsp衍生物、脂连蛋白及其衍生物、由多肽构成的clsp或该clsp衍生物所具有的clsp活性的增强或保护剂、以及构成融合蛋白的多肽还仅称为“本发明(的)多肽”。
[0091]
关于本发明多肽,为了确定2个氨基酸序列中的序列的同一性,对序列进行预处理以达到比较最适的状态。例如,通过在一个序列中插入缺口,进行与另一个序列的比对的最优化。之后,比较各位点的氨基酸残基或碱基。在第一序列中的某个位点存在与第二序列的
相当位点相同的氨基酸残基或碱基的情况下,这些序列在其位点上是相同的。2个序列中的同一性以序列间的相同位点数与总位点(总氨基酸或总碱基)数的百分率表示。
[0092]
按照上述原理,2个氨基酸序列中的同一性可按照本领域技术人员已知的任意方法确定。例如,可根据karlin和altshul的算法(proc. natl. acad. sci. usa 87:2264-2268, 1990和proc. natl. acad. sci. usa 90:5873-5877, 1993)确定。采用了这种算法的blast程序由altshul等人开发(j. mol. biol. 215:403-410, 1990)。
[0093]
而且,gapped blast是以高于blast的灵敏度确定同一性的程序(nucleic acids res. 25: 3389-3402, 1997)。上述程序主要用于从数据库中检索相对于所提供的序列显示高度同一性的序列。这些例如可在美国国家生物技术信息中心(national center for biotechnology information)的互联网上的网站中利用。
[0094]
或者,作为序列间的同一性,还可采用使用由tatiana a. tatusova等人开发的blast 2 sequences软件(fems microbiol lett., 174:247-250, 1999)确定的值。该软件可在美国国家生物技术信息中心的互联网上的网站中利用,也可获取。使用的程序和参数如下。在氨基酸序列的情况下,作为使用blastp程序的参数,使用open gap:11和extension gap:1 penalties,gap x_dropoff:50,expect:10,word size:3,filter:on。而且,还可使用高灵敏度的fasta软件(w. r. pearson和d. j. lipman, proc. natl. acad. sci. usa, 85: 2444-2448, 1988)从数据库中检索显示同一性的序列。所有参数也均在网站上用作默认值。
[0095]
上述的本发明多肽也可具有利用已知方法通过修饰、添加、突变、取代或删除等改变而成的形态。这样的官能团的改变可利用本领域技术人员已知的任意方法,例如为了保护多肽、控制多肽的稳定性或组织转移性、或控制多肽的活性等而进行。
[0096]
即,本发明多肽可通过翻译后修饰等进行天然修饰。另外,也可进行人工修饰。修饰包括:肽的主链、氨基酸侧链、氨基末端、或羧基末端等的修饰。另外,多肽可以分支,也可以是环状。修饰包括:乙酰化、酰基化、adp核糖基化、酰胺化、[黄素(flavin)、核苷酸、核苷酸衍生物、脂质、脂质衍生物、或磷脂酰肌醇]等的共价键、交联形成、环状化、二硫键形成、脱甲基化、焦谷氨酸化、羧基化、糖基化、羟基化、碘化、甲基化、豆蔻酰化、氧化、磷酸化、泛素化等,但并不限于这些。而且,上述肽或多肽也可形成本领域技术人员已知的任意的盐和酯体。
[0097]
而且,本发明的多肽也可与已知的任意的向神经肽形成融合多肽,这样的融合多肽可按照本领域技术人员已知的任意方法容易地合成。
[0098]
本发明的多肽可根据本领域技术人员已知的关于clsp和脂连蛋白等的基因或氨基酸序列信息,由来自人和小鼠等适当种的细胞株等调制,而且,可利用已知的肽合成技术进行制造。另外,也可利用本领域技术人员已知的基因工程学方法,通过将包含编码它们的dna的载体等引入到适当的宿主细胞等中使其表达来制造。此时,例如在clsp衍生物、或脂连蛋白衍生物的情况下,可通过本领域技术人员已知的方法/手段适当改变其一部分氨基酸序列来调制。
[0099]
这样的载体是质粒或病毒性载体等本领域技术人员已知的任意形态,可按照本领域技术人员已知的任意方法容易地调制。如此操作而得的载体除包含本发明的位点特异性重组酶的编码区以外,还在5’和3’适当包含非编码序列(包括核转移信号、标签序列、非转
录序列、非翻译序列、启动子、增强子、抑制子、转录因子结合序列、剪接序列、聚a (poly a)添加序列、ires、mrna稳定化/不稳定化序列等),作为表达载体起作用。
[0100]
通过使用了这样的载体的本领域技术人员已知的任意方法、例如脂质转染法、磷酸钙法、以及电穿孔和粒子枪等各种物理方法,可容易地转化适当的宿主细胞。
[0101]
对宿主细胞没有特别限定,例如可使用包括人、猴和小鼠等的哺乳动物细胞、植物细胞、昆虫细胞、和大肠杆菌等细菌类。如此制作的转化细胞可在本领域技术人员已知的任意条件下进行培养,由培养的菌体或其培养上清等适当组分容易地调制作为目标的本发明的多肽等。
[0102]
本发明多肽可用作:用于抑制与阿尔茨海默病相关的神经细胞的功能障碍或神经细胞死亡的药物组合物、例如用于预防或治疗伴有与阿尔茨海默病相关的记忆损伤或神经变性的疾病的药物组合物的有效成分。
[0103]
而且,使用本发明多肽,除阿尔茨海默病以外,还可预防/治疗伴有记忆损伤或神经变性的疾病、例如因脑缺血引起神经细胞的细胞死亡而引发的疾病(t. kirino, 1982, brain res., 239: 57-69)。此外,伴有痴呆的帕金森病(m. h. polymeropoulos等人, 1997, science, 276: 2045-2047)、弥漫性路易式小体(lewy bodies)病(m. g. spillantini等人, 1998, proc. natl. acad. sci. usa, 95: 6469-6473)、唐氏综合征所伴随的痴呆等也成为治疗或预防的对象。另外,从作为app的类似分子的aplp1被视为先天性肾病综合征的病源基因(lenkkeri, u等人, 1998, hum. genet. 102: 192-196)的角度考虑,肾病综合征等肾病也成为治疗或预防的对象。
[0104]
本发明的药物组合物除了可直接对患者给予有效成分本身以外,也可利用已知的制剂学方法制成制剂。例如,考虑与药理学上可接受的载体或介质、具体而言与灭菌水或生理盐水、植物油、乳化剂、悬浮剂、表面活性剂、稳定剂、缓释剂等适当组合制成制剂后给药。本发明的药物组合物可以是水溶液、片剂、胶囊、锭剂、口含片、酏剂、悬浮液、糖浆、滴鼻液或吸入液等形态。作为有效成分的肽或多肽的含量可根据使用目的和制剂形态等适当确定。
[0105]
对患者的给药可根据有效成分的性质,例如以经皮、鼻腔内、经支气管、肌肉内、腹腔内、静脉内、脊髄腔内、脑室内或口服的方式进行,但并不限于这些。在用于治疗脑神经变性疾病的情况下,希望本发明的药物组合物是通过包括静脉内、脊髄腔内、脑室内或硬脑膜内注射的任意的适当途径导入至中枢神经系统。给药量、给药方法根据本发明的药物组合物的有效成分的组织转移性、治疗目的、患者的体重或年龄、症状等而变化,但本领域技术人员可适当选择。例如,1天1次~多次,每1次处置可在适当期间内给予数十μl左右的药剂。有效成分例如可设为10pmol~100nmol左右的范围的浓度。
[0106]
如此,本发明的药物组合物可广泛用于预防或治疗阿尔茨海默病等伴有神经细胞的细胞功能障碍或神经细胞死亡的疾病、或者伴有记忆损伤或神经变性的疾病。
[0107]
因此,本发明涉及:抑制神经细胞的功能障碍或细胞死亡的方法,包括使本发明的多肽与神经细胞接触的工序;治疗阿尔茨海默病等伴有神经细胞的细胞功能障碍或神经细胞死亡的疾病、或伴有记忆损伤或神经变性的病症的方法,包括对患有或疑似患有该疾病或病症的人等动物对象(个体)给予本发明的药物组合物的阶段;以及治疗伴有神经变性障碍的疾病的方法。
[0108]
本发明还涉及:检测本发明的多肽对神经细胞的功能障碍或细胞死亡的抑制活性的方法,该方法包括:工序(a),在clsp的阻碍剂的存在/不存在下、和该多肽的存在/不存在下,诱导神经细胞的功能障碍或细胞死亡;工序(b),检测神经细胞的细胞功能障碍或细胞死亡;以及工序(c),比较在该多肽的存在/不存在下的神经细胞的功能障碍或神经细胞死亡等。
[0109]
根据上述的方法,可检测:本发明的多肽对神经细胞的功能障碍或细胞死亡的抑制活性、以及增强或保护clsp或clsp衍生物所具有的clsp活性免受clsp阻碍剂的阻碍的活性。
[0110]
具体的操作例如可按照本说明书中记载的方法来进行。该方法可用于确定本发明的多肽是否对各种细胞中的细胞死亡具有抑制效果、或者定量其抑制效果。对细胞没有特别限定,使用可引起细胞死亡的各种细胞。另外,细胞死亡的诱导可根据各自的细胞使用已知的细胞死亡诱导系统。另外,使用神经细胞,还可用于检测本发明的多肽等对诱导神经细胞死亡的各种刺激、环境变化、或基因表达等各种条件的效果。另外,这样的检测可用于检测生物种或亚种或个体间可存在的在神经细胞死亡中对本发明的多肽等的敏感性的不同。由此,例如可在民族、人种或个人间研究本发明的多肽的有效性。利用这样的方法,例如可进行面向临床应用的详细的条件研究。
[0111]
另外,本发明还涉及:筛选调节本发明的多肽或clsp对神经细胞的功能障碍或神经细胞死亡的抑制活性的物质(受试物质)的方法。该方法可用于测定该受试物质对本发明的多肽或clsp所产生的对神经细胞的功能障碍或神经细胞死亡的抑制活性的效果(影响)。认为本发明的多肽或clsp与神经细胞表面作用以发挥细胞死亡抑制效果。如果采用该方法,则可验证:可阻碍这些多肽与细胞表面接触的候选化合物或反之可促进该接触的候选化合物的作用。
[0112]
该筛选方法包括以下工序:工序(a),在本发明的多肽或clsp的存在下、在受试物质的有无下诱导神经细胞的功能障碍或神经细胞死亡;工序(b),检测神经细胞的功能障碍或神经细胞死亡;以及工序(c),选择调节本发明的多肽或clsp对神经细胞的功能障碍或神经细胞死亡的抑制活性的物质。在工序(c)中,可与任意的对照中的情况作对比。例如,在工序(c)中可选择与在受试物质的不存在下进行检测的情况相比在受试物质的存在下促进或抑制神经细胞的功能障碍或神经细胞死亡的化合物。促进神经细胞的功能障碍或神经细胞死亡的化合物成为阻碍本发明的多肽或clsp的作用的化合物的候选,进一步抑制神经细胞死亡的化合物成为进一步促进本发明的多肽或clsp的作用的化合物的候选。
[0113]
另外,在上述的筛选中,也可将受试物质以外的其他化合物的情况作为对照。例如,在工序(b)中也可使用可调节本发明的多肽或clsp对神经细胞的功能障碍或神经细胞死亡的抑制的其他化合物进行检测,在工序(c)中也可选择与在该化合物的存在下的情况相比在工序(a)中使用的受试物质的存在下更促进或抑制神经细胞的功能障碍或神经细胞死亡的化合物。在这样的筛选中,可筛选在本发明的多肽或clsp对神经细胞的功能障碍或神经细胞死亡的抑制的调节能力方面具有较现有化合物更高的效果的化合物。
[0114]
作为用于上述筛选的受试物质,例如可列举:纯化蛋白(包括抗体)、基因文库的表达产物、合成肽的文库、细胞提取液、细胞培养上清、合成低分子化合物的文库、土壤等天然材料、放线菌肉汤等含有细菌释放物质的溶液等,但并不限于这些。神经细胞死亡的诱导或
本发明的多肽的给药可按照本领域技术人员已知的任意方法来进行。
[0115]
对将受试物质适用于细胞的时期没有特别限定,可在适用本发明的多肽之前、之后或同时适用。对受试样品的适用方法没有限定,如果是培养细胞系统,例如可添加在培养基中。另外,如果是核酸,则可引入到细胞内。可通过其他任意的给药方法适用受试样品。
[0116]
通过检查上述化合物的作用而评价的物质、或通过筛选得到的物质成为调节本发明的多肽的活性的化合物的候选,考虑将其应用于预防或治疗包括阿尔茨海默病在内的疾病。
[0117]
本发明还涉及:与本发明的多肽结合的物质(化合物)的筛选方法,该方法包括:工序(a),使受试物质与该多肽接触;工序(b),检测该多肽等与受试物质的结合活性;以及工序(c),选择具有与该多肽结合的活性的物质。
[0118]
本发明的多肽根据筛选方法可作为可溶性多肽、且以与载体结合的形式用于筛选。本发明的多肽可被标记。作为标记,可列举:放射性同位素标记、荧光物质标记、生物素或洋地黄毒苷标记、标签序列的添加等。
[0119]
作为用于筛选的受试物质,例如可列举:纯化蛋白(包括抗体)、基因文库的表达产物、合成肽的文库、细胞提取液、细胞培养上清、合成低分子化合物的文库、土壤等天然材料、放线菌肉汤等包含细菌释放物质的溶液等,但并不限于这些。受试物质根据需要进行适当标记后使用。作为标记,例如可列举:放射标记、荧光标记等,但并不限于这些。
[0120]
例如,在筛选与本发明的多肽结合的蛋白的情况下,在固定有本发明的多肽的亲和柱上加载预测正在表达与本发明的多肽结合的蛋白的组织或细胞的细胞提取物,并纯化特异性地与柱结合的蛋白,从而可实施与本发明的多肽结合的蛋白的筛选。
[0121]
再由预测正在表达与本发明的多肽结合的蛋白的组织或细胞(例如脑皮质组织、或f11等神经细胞)制作使用了噬菌体载体的cdna文库,在琼脂糖上形成噬斑,使用已标记的本发明的多肽等通过蛋白质印迹法进行筛选,或者也可按照下述的“双杂交系统”等实施:使gal4 dna结合区等dna结合肽和gal4转录活化区等转录活化肽分别作为本发明的多肽等与受试蛋白的融合蛋白表达,通过连接在具有dna结合肽的结合序列的启动子的下游的报道基因的表达,检测本发明的多肽等与受试蛋白的结合。
[0122]
还考虑通过本发明的筛选来筛选本发明的多肽的受体。这种情况下,受试样品优选由预测正在表达受体的组织或细胞、例如脑皮质组织、神经细胞株、或成神经细胞瘤或畸胎瘤细胞等调制。作为神经细胞株,例如可列举:f11细胞、pc12细胞(l. a. greene和a. s. tischler, 1976, proc. natl. acad. sci. usa, 73:2424-2428)、ntera2细胞(j. skowronski和m. f. singer, 1985, proc. natl. acad. sci. usa, 82:6050-6054)、sh-sy5y细胞(l. odelstad等人, 1981, brain res., 224:69-82)等。
[0123]
另外,还考虑使合成化合物、天然物库或随机噬菌体肽展示文库等与固定化的本发明的多肽作用,筛选结合的分子。另外,也可通过利用表面等离子体共振现象的结合的检测进行筛选(例如biacore公司制造等)。这些筛选也可通过利用组合化学技术的高通量筛选来进行。
[0124]
通过本发明的筛选得到的与本发明的多肽结合的化合物成为调节本发明的多肽活性的化合物的候选,考虑将其应用于预防或治疗包括阿尔茨海默病在内的疾病。
[0125]
以下,根据实施例进一步具体地说明本发明。尚需说明的是,本发明的技术范围并
不受这些记载的任何限制。
实施例
[0126]
[多个clsp相互作用因子的鉴定]在以前的研究中14-3-3σ、14-3-3β、钙网蛋白、erp27、核仁蛋白、膜联蛋白ii和膜联蛋白v等多个蛋白被鉴定为推测的clsp结合因子(15)。这些之中,选择据报道分泌至细胞外空间的蛋白用于本发明中的分析。另外,在本发明中新发现了载脂蛋白e (apoe)和脂连蛋白与clsp结合(图1)。
[0127]
[载脂蛋白e、14-3-3蛋白和钙网蛋白为clsp的阻碍物质]如以上所示(5),v642i-淀粉样β前体蛋白(v642i-app)的过度表达会引起sh-sy5y成神经细胞瘤细胞死亡,但通过与细菌中产生的重组clsp (500pm或1nm)同时培养,v642i-app诱导性神经元死亡被完全抑制(图2a)。根据剂量反应性分析,推测细菌中产生的clsp的50%有效浓度为约200pm (图2a),显示较哺乳动物细胞中产生的重组clsp的50%有效浓度稍大(5)。有趣的是,在培养基中添加重组载脂蛋白e3或e4 (apoe3或apoe4)会剂量应答性地阻碍由clsp介导的对v642i-app诱导性神经元死亡的保护(图2b和c)。apoe3在5nm的浓度下完全阻碍了1nm的clsp的效果(图2b),另一方面,apoe4在1nm的浓度下完全阻碍了1nm的clsp的效果(图2c)。该结果显示:apoe4的阻碍效果较apoe3稍强。同样,通过与重组14-3-3σ同时培养,由clsp介导的对v642i-app诱导性神经元死亡的保护受到阻碍(图3a)。在10nm的浓度下开始观察到14-3-3σ对10nm的clsp的阻碍效果,然后在该特定的实验中添加20nm的重组14-3-3σ时获得了完全阻碍。其他的14-3-3蛋白也阻碍了由clsp介导的对v642i-app诱导性神经细胞死亡的阻碍,在50nm的重组14-3-3σ时获得了完全阻碍(图3b~e)。同样,钙网蛋白在50nm的浓度下完全阻碍了10nm clsp的效果(图3f)。因此,这些结果证实了clsp阻碍剂在与培养基中的clsp浓度同等或5倍以上的浓度下显示完全的clsp阻碍效果。对照而言,膜联蛋白ii、膜联蛋白iv或脂连蛋白即使在高达培养基中的clsp浓度的5倍或10倍的浓度下也没有显示阻碍活性(图3f和图s-1)。
[0128]
[即使在浓度非常高的clsp阻碍物质的存在下脂连蛋白也维持clsp活性]推测人脑脊液(csf)中的clsp的浓度为3~6nm (14)。已知apoe由星形胶质细胞和小胶质细胞产生,在人cns中有相当比例的apoe与脂质和其他载脂蛋白同时被动员形成高密度脂蛋白样脂蛋白(16、17)。推测人csf中的apoe的浓度为40~200nm (18-20)。另一方面,推测人csf中的14-3-3σ的浓度远低于1nm (参照图s2)。另外,根据以前的研究推测人csf中的14-3-3γ的浓度低于1nm (21)。而且,推测人血清中的钙网蛋白的浓度约为10pm左右(22),但目前为止还未测定人csf中的浓度。综合这些见解,作为整体量的apoe的浓度超过完全阻碍clsp功能所需的浓度的10倍以上。另一方面,其他的阻碍物质的浓度很可能不足以阻碍人cns中的clsp。
[0129]
如上所述,由于在人cns中存在非常大量的clsp阻碍物质(主要由apoe构成),因此clsp活性在体内看起来为零。然而,根据以前的研究(35),自然会认为至少在正常人中存在clsp活性。因此,接下来研究了其他clsp结合物质中的哪一种在保护clsp免受clsp阻碍物质的影响以保持活性的假设。因此,在包含1nm clsp和10nm apoe3的细胞死亡测定系统中以与apoe3的浓度相等的浓度添加作为候选的膜联蛋白ii、膜联蛋白v或脂连蛋白的重组蛋
白。其结果,脂连蛋白使apoe3介导的对clsp活性的阻碍完全失效(clsp保护活性) (图4a)。另一方面,在试验的浓度下膜联蛋白ii和膜联蛋白v均未显示这样的中和活性(图4b和c)。为了确定脂连蛋白的最小失效浓度,接下来阶段性地减少脂连蛋白的浓度。其结果,脂连蛋白按以下的浓度比完全抑制apoe4的clsp阻碍活性(clsp、1nm;apoe4、10nm:脂连蛋白、1nm) (图5a)。而且,即使进一步降低浓度至100pm的浓度,脂连蛋白也部分地抑制了apoe4的clsp抑制活性(clsp、1nm:apoe4、10nm:脂连蛋白、100pm)。另一方面,反之即使将apoe4的浓度增加至50nm,1nm的脂连蛋白的保护效果也完全没有减弱(图5b)。这些结果显示:即使在绝对高的浓度的apoe4的存在下,1nm的脂连蛋白也将活性型clsp的浓度维持在100%有效的水平。野生型脂连蛋白在生物体内自发地多聚体化,形成3种多聚体型。而且,3聚体被称为低分子量脂连蛋白,6聚体被称为中分子量脂连蛋白,而8聚体或其以上等被称为高分子量脂连蛋白(23)。根据以前的研究,已知中分子量或高分子量的脂连蛋白在由普通脂连蛋白受体介导的代谢调节活性中起到核心作用(23)。在该研究中,使用未形成中分子量或高分子量的脂连蛋白的重组三聚体脂连蛋白,发现三聚体脂连蛋白具有与野生型脂连蛋白同样的clsp增强效果(图s3)。该结果意味着:脂连蛋白的中分子量或高分子量多聚体化并不是脂连蛋白的clsp保护效果所必须的。根据以前的研究可知:关于由脂连蛋白的普通受体介导的代谢调节功能,中分子量或高分子量多聚体化的脂连蛋白显示更高的活性(23)。总之,这里发现的脂连蛋白的clsp保护活性强力支持不是普通脂连蛋白受体介导的效果的可能性。进一步对apoe以外的clsp阻碍物质也进行同样的研究,发现脂连蛋白对14-3-3σ或钙网蛋白的clsp阻碍效果也完全显示保护活性(clsp、1nm:14-3-3σ或钙网蛋白、2nm或10nm:脂连蛋白、1nm) (图6)。
[0130]
[脂连蛋白增强clsp活性]而且,还发现了:脂连蛋白除了具有上述的clsp保护效果,还具有增强clsp活性本身的效果。如图2a所示,clsp在50pm的浓度下对v642i-app诱导细胞死亡没有显示抑制活性。然而,在200pm的脂连蛋白的存在下,clsp在50pm或25pm的浓度下分别显示几乎完全的或一部分的细胞死亡抑制活性(图7a)。该结果显示:脂连蛋白通过与clsp结合而增强clsp活性。本发明人进一步发现:即使在同时给予的脂连蛋白的浓度减少至100pm的情况下,脂连蛋白也对50pm浓度的clsp显示一部分增强活性(图7b)。综合这些结果显示:用于对单独时不具活性的50pm的clsp赋予完全的细胞死亡抑制活性的最小脂连蛋白浓度为200~250pm。
[0131]
[脂连蛋白与clsp的结合的解离常数和载脂蛋白e4与clsp的结合的解离常数近似]如上所示,显示脂连蛋白使50倍高的浓度的apoe的clsp活性阻碍效果完全失效(保护效果) (图4和图5)。赋予脂连蛋白的这种强效的clsp保护效果可通过如下所示的两种机制来说明。第一种情况:脂连蛋白和apoe竞争性地与clsp的相同位点结合,并且,脂连蛋白与clsp之间的结合亲和性远强于载脂蛋白e与clsp之间的结合亲和性(竞争性拮抗剂)。第二种情况:脂连蛋白与不同于apoe结合区的clsp区结合,从而通过单独结合使clsp活性上升,在同时存在clsp阻碍物质的结合的情况下,抑制其阻碍效果以保持clsp活性(非竞争性拮抗剂)。
[0132]
研究了这2种机制中的哪一种是正确的。首先,为了查明apoe3或apoe4的存在对
clsp和脂连蛋白或膜联蛋白ii会产生怎样的影响、或者其反之如何,在共价键合有clsp的sepharose 4b珠粒(clsp珠粒)中混合该蛋白,进行pull-down测定(共沉淀实验),研究与clsp的结合(图8a)。作为条件,混合物中的clsp、脂连蛋白、膜联蛋白ii、apoe3或apoe4的浓度均设为1nm。首先,在不存在其他蛋白的情况下,一定量的脂连蛋白、膜联蛋白ii、apoe3或apoe4与clsp珠粒共沉淀(泳道2~4和7)。接下来,若与脂连蛋白一同共沉淀则稍有减少,但仍有相当量的apoe3或apoe4与clsp珠粒共沉淀(泳道5和8)。同样,若与apoe3或apoe4一同共沉淀则稍有减少,但依然有相当量的脂连蛋白共沉淀(泳道5和8)。重要的是,共沉淀apoe的量与共沉淀脂连蛋白的量相等或比其稍多(图10a)。另一方面,即使加入膜联蛋白ii,也没有减少与clsp共沉淀的apoe3或apoe4的沉淀量(泳道6和9)。反之,在apoe3或apoe4的存在下,膜联蛋白ii几乎没有与clsp珠粒共沉淀(泳道6和9)。这些结果显示:关于脂连蛋白与clsp的结合,并不是通过与apoe竞争来抑制apoe的阻碍作用(第一可能性的否定)。
[0133]
接下来,测定关于clsp与脂连蛋白之间、以及clsp与apoe4之间的结合的解离常数(kd) (图8b和c)。为了该目的,将脂连蛋白或apoe4蛋白与96孔板缀合。将c末端用作为产生化学发光的标签的hibit标记的各种浓度的重组clsp添加至板上以进行同时培养。洗涤后,测定孔上的与脂连蛋白或apoe4结合的clsp-hibit的量。通过斯卡查德分析测定脂连蛋白与clsp之间、或者apoe4与clsp之间的解离常数分别为8.8或7.8pm (图8c)。两者近似的这个结果完全否定了上述的第1种可能性。
[0134]
[脂连蛋白和载脂蛋白e4与clsp的不同亚结构域结合]为了探究第2种可能性,制作apoe4-mychis和脂连蛋白-mychis,与以前构建的重组野生型clsp或clsp缺失突变体(5、图9a)混合,进行pull-down测定(图9)。其结果,仅由her构成的突变体没有与apoe4共沉淀(结合),而另外的4个clsp突变体与apoe4结合。另一方面,仅δn2与脂连蛋白结合,而其他的4个突变体没有与其结合(图9b和c)。这些结果意味着:clsp中的apoe4结合位点位于ehr的外侧,脂连蛋白结合位点包含ehr。因此判明:脂连蛋白通过与ehr结合来增强和保护clsp活性;而且,一旦脂连蛋白与ehr结合,则经由非ehr区引起的clsp阻碍物质的阻碍效果被消除。
[0135]
通过进一步进行同样的pull-down实验,发现脂连蛋白与由氨基酸1-61构成的clsp的n末端区结合,相对于此,apoe4没有与其结合(图s4a和b)。若与图9的结果相结合,则该结果意味着:apoe与clsp的c末端区(氨基酸62~146)结合。另外,通过其他的类似实验,本发明人还发现:clsp与位于脂连蛋白的中央部分的脂连蛋白的所谓“胶原同源区(adncol)”结合(图s4c)。
[0136]
[在ad患者中csf中的脂连蛋白浓度显著降低]为了推测人cns中的脂连蛋白浓度,使用脂连蛋白elisa测定试剂盒,测定由尸检ad患者和非ad病例得到的csf中的脂连蛋白的水平(表1、表s1和图10)。其结果,发现在ad患者中csf脂连蛋白的水平较非ad病例低(图10b、表1)。ad患者中的csf脂连蛋白的平均
±
sem浓度为0.31
±
0.13nm,而非ad病例中的该值为0.96
±
0.19nm (非配对t检验、p=0.0065)。该结果暗示:与以前的研究(30)结果基本一致,在ad患者的csf中脂连蛋白水平显著降低。几个非ad病例的脂连蛋白浓度显著低于非ad病例的平均,与ad患者的平均水平几乎相等。
[0137]
然而,ad患者的平均年龄(78.5
±
0.9岁)显著小于非ad病例的平均年龄(86.3
±
1.4岁以上) (非配对t检验;在将“大于”看作“等于”的情况下p《0.0001) (参照表s1)。因
此,与ad的存在相比,年龄更有可能对csf脂连蛋白的浓度产生影响。为了研究该可能性,从表1的所有病例中选择年龄为81~88岁的病例(ad的n=6、平均年龄
±
sem=83.0
±
0.6岁;非ad的n=5、平均年龄
±
sem = 85.2
±
1.2岁;针对年龄的非配对t检验;p=0.11) (表2),比较它们的csf脂连蛋白水平。其结果,发现即使在该年龄团体中在ad患者的csf中脂连蛋白水平也显著下调(ad、0.30
±
0.07nm;非ad、1.41
±
0.16nm;非配对t检验、p《0.0001) (图10c、表2)。进一步利用来自所有受试者的数据研究csf脂连蛋白水平与年龄是否相关。不出所料,在年龄与csf脂连蛋白浓度之间没有显著的相关(相关系数=0.0055) (图s5)。
[0138]
[在ad患者的神经元中作为人体肽/clsp诱导性细胞内信号传递路径的核心效应子的sh3bp5降低]根据图12的结果,显示在ad患者的脑中相当于活性型clsp的量的clsp /脂连蛋白复合体的量少于非ad病例的该量。为了获得该实验事实的确认(内情),尝试通过测定神经元内sh3bp5的水平将神经元内的clsp诱导性信号传递的强度定量化。作为此目的的方法,测定sh3bp5的依据在于:通过以前的研究,证实了sh3bp5是由hthnr介导的人体肽/clsp诱导性细胞内信号传递的核心效应子,若人体肽/clsp与hthnr结合,则表达水平上升(24)。实际上,在该研究(24)中,若clsp/人体肽与hthnr结合,则经由stat3活化sh3bp5的转录。其结果,显示sh3bp5表达水平上升,高水平的sh3bp5直接与jnk形成复合体以阻碍jnk,从而阻碍v642i-app诱导性死亡信号。为了该目的,首先,使用sh3bp5抗体对由尸检ad患者和肌萎缩性侧索硬化症(als)患者得到的脑的颞叶或枕叶(非运动神经元区)进行免疫组织化学染色(表3和表s2)。使用als患者的脑作为阴性对照的原因在于:在als中仅在颞叶的运动区的运动神经元中发生神经变性,在颞叶或枕叶的神经元中未见异常。作为该实验的结果,发现与als相比在ad的皮质的神经元中sh3bp5的水平降低(非配对t检验、p=0.0256) (图11a、b和c、表3)。考虑到ad患者的平均年龄高于als患者的平均年龄(78岁对69岁),我们还研究了与ad的存在相比是否年龄才是sh3bp5水平的决定性因素,其结果,发现高龄者(71岁以上)的sh3bp5水平并非显著低于低龄者(小于71岁) (非配对t检验、p=0.633) (图s6)。
[0139]
另外,采用sh3bp5 elisa测定,测定来自尸检ad患者和非ad病例的颞叶的组织裂解物中的sh3bp5水平(表4和表s3)。其结果,发现ad患者颞叶中的sh3bp5水平显著低于非ad病例(非配对t检验、p=0.0084) (图11d和e、表4)。
[0140]
作为进一步的展开,发现作为clsp衍生物的clsp1-61没有与apoe4结合(图s4a和b)。而且,由于包含her,所以很有可能保持clsp活性。在采用细胞死亡测定来研究其细胞死亡抑制活性时,最小细胞死亡抑制浓度为0.5nm (图l1),与野生型clsp几乎相同(图2)。进一步研究3种clsp阻碍物质能否抑制clsp1-61的活性时,不出所料,即使加入10倍量的各阻碍物质(apoe4、14-3-3σ、钙网蛋白),也无法阻碍clsp1-61的活性(图l2)。因此,判明clsp1-61是不受阻碍物质的阻碍、且维持活性的clsp衍生物。
[0141]
另外,发现clsp与脂连蛋白的胶原区(adncol)结合(图s4c)。因此,研究了脂连蛋白的clsp活性增强效果是否仅通过adncol就足够。其结果,1nm的adncol与1nm的野生型脂连蛋白相同,增强50pm的clsp的活性,完全抑制细胞死亡(图l3)。进一步运用相同的测定,增减adncol的量进行研究,求出对50pm的clsp赋予完全的细胞死亡抑制活性的最小adncol浓度。其结果,判明clsp增强活性稍弱于野生型脂连蛋白的该活性,浓度为0.5nm (图l4)。因此,判明adncol是活性稍弱于野生型脂连蛋白、但具有几乎相同的作用的蛋白。
[0142]
[由clsp1-61和脂连蛋白的胶原同源区构成的融合多肽具有强效的ad保护活性]接下来,调制由clsp1-61和脂连蛋白的胶原同源区、以及由野生型clsp和脂连蛋白的胶原同源区构成的2种杂交多肽(分别称为“clspcol”和“wt-clspcol”),研究它们是否保持clsp和脂连蛋白两者的活性。这两种杂交多肽的区域通过编码myc标签和hisg标签(由6
×
组氨酸和甘氨酸构成)的肽连接。与clsp1-61和野生型clsp (图l1)相比,clspcol和wt-clspcol具有更强效的clsp活性(完全抑制神经细胞死亡的两种肽的最小浓度均为约100pm:图l5)。其结果显示:clsp1-61和野生型clsp的功能不会被脂连蛋白的胶原同源区的c末端键破坏,相反,脂连蛋白的胶原同源区增强了clsp1-61和野生型clsp的功能。因此,认为脂连蛋白的胶原同源区的功能不会被clsp1-61的n末端键破坏。
[0143]
[clspcol有效地透过血脑屏障]在小鼠中,在腹腔内注射5nmol的clsp后1小时,脑脊液和血清中的clsp的浓度为约5nm和500nm (5)。另一方面,人脑脊液中的脂连蛋白浓度为人血清中的脂连蛋白浓度的1/1000 (30)。因此,研究clspcol和wt-clspcol是否以与小鼠的野生型clsp同等的速度透过血脑屏障。在腹腔内注射10nmol的clspcol后1小时,血清和含间质液(isf)的脑裂解物中的clspcol浓度为约305nm和72nm (图l6和表1)。另一方面,注射后1小时的血清和含间质液的脑裂解物中的wt-clspcol的推测浓度为约53nm和2.1nm。含isf的脑裂解物中的wt-clspcol的浓度小于所采用的elisa测定的最低检测限(4.5nm),因此也许暂定浓度2.1nm并不正确,但其浓度确实小于4.5nm。推测脑裂解物中的clspcol的浓度为血清中的浓度的约1/4~1/5,另一方面,推测wt-clspcol的浓度小于血清中的该浓度的1/10。因此,clspcol的中枢神经系统转移也比wt-clspcol的该转移有效。另外,考虑到所发表的结果中的clsp从血清到脑脊液的转移效率(5),认为透过血脑屏障的clspcol的转移远较wt-clsp有效。
[0144]
[clspcol不被apoe3和14-3-3σ阻碍但被钙网蛋白轻度阻碍]在研究clspcol是否被3种阻碍剂阻碍时判明:虽然不被10倍高的浓度的apoe3和14-3-3σ阻碍,但出乎意料被10倍高的浓度的钙网蛋白轻度阻碍(图x1)。另一方面,wt-clspcol不被所有的阻碍剂阻碍(图x2)。在改变所添加的钙网蛋白的浓度进行详细研究时判明:钙网蛋白从1nm的浓度起开始对100pm的clspcol显示阻碍活性(图x2)。
[0145]
[考察]在包含ad在内的神经变性疾病中,神经变性是由增加的神经毒性物质或神经障碍机制引起的这种想法通常被广泛接受。在ad中,在20年或更长时间内就将aβ (老人斑中的凝集的原纤维型aβ和/或可溶性aβ寡聚物)水平上升视为主要的损伤原因(2)。此外,还显示了过度磷酸化tau、以及与aβ水平上升没有直接关系的淀粉样β前体蛋白和早老蛋白(presenilin)的相关的神经障碍机制作为毒性参与的可能性。另外,通过先前研究或本研究提示:除已知的这些神经障碍机制以外,ad保护因子的降低/减弱也可能有助于ad的恶化。其中,推测clsp作为该ad保护(防御)因子起到核心作用的可能性高(6)。
[0146]
人体肽和clsp在体外阻碍与ad相关的神经细胞死亡(5、6)。另外,在ad模型小鼠中clsp抑制突触消失和记忆障碍(8)而与aβ的调节无关。后者的结果还被一系列的先前研究(25-27)所支持。即,这是作为hthnr的其他激动剂的人体肽的强效衍生物抑制ad模型小鼠中的记忆障碍的研究。根据这些结果,本发明人提出了关于ad病因的假设:2种现象、即ad相关神经毒性的增加和ad保护活性的减少这两者是ad发病所必需的。若认为该假设正确,则
在足够浓度的活性clsp的存在下,即使ad相关神经毒性充分增强,也无法引起神经细胞死亡(和功能不全),因此不会患上ad。另外,在没有足够的ad相关神经毒性的情况下,即使clsp效果降低,也不会引起神经细胞死亡(和功能不全) (即,不会患上ad)。在本研究所示的数据中,除了几乎所有的ad病例,还有几例“非ad”对照的cns中的脂连蛋白和sh3bp5水平降低(图10和11),这一实验结果表明:这些想法是极为妥当的。
[0147]
认为clsp是与异源三聚体人体肽受体结合、并活化stat3诱导性存活信号传递路径的核心ad保护因子(5、6和8),它的异常调节很有可能有助于ad的病因。在通过本发明确认的多个clsp阻碍物质中,若考虑浓度和活性,则认为apoe是核心阻碍物质(图2)。推测在人cns中总apoe的浓度绝对高于clsp的浓度(18-20) (14)。因此,如果仅由clsp和非常大量的clsp阻碍剂构成的体内clsp活性调节模型是正确的,则ad保护活性在体内几乎为零的可能性高。
[0148]
然而,如本发明所示,如果脂连蛋白是通过与clsp的内源性人体肽同源区(ehr)结合来增强clsp活性、进而以优先的形式保护clsp免受clsp阻碍剂的影响(图5~7),则无论存在怎样高浓度的阻碍物质,体内的该clsp活性也会得到担保。即使在高得多的浓度的clsp阻碍剂的存在下,0.2-0.25nm的脂连蛋白也可完全维持1nm的clsp活性(图5和7)。在非ad的情况下,csf中的脂连蛋白浓度为0.96
±
0.19nm (图10和表1),其结果,维持clsp活性的可能性高。
[0149]
脂连蛋白在外周组织中发挥包括葡萄糖和脂质代谢在内的各种代谢功能(28)。其或许是经由细胞膜上的2种普通的脂连蛋白受体增加胰岛素信号传递、抗炎性、抗氧化性和抗粥样斑块产生性的功能。认为透过血脑屏障的脂连蛋白的移动非常有限。csf中的脂连蛋白浓度较血清中的浓度几乎低103倍(29、30)。考虑到在cns中存在普通的脂连蛋白受体,假定脂连蛋白在cns中作为葡萄糖代谢的调节因子、神经新生促进剂起作用,进而例如作为对缺血性脑损伤的保护因子起作用(31)。大量的研究提供了以下的证据:脂连蛋白的缺乏或脂连蛋白信号传递的异常调节与ad的发病有关(31)。血清脂连蛋白水平的上升(29、30)有可能是ad的独立的危险因子(32)。另一方面,在某研究中显示:在具有低血清脂连蛋白浓度的ii型糖尿病患者中显著多地出现ad样症状(33)。在ad患者的csf中脂连蛋白水平下调,与aβ水平的增加逆相关(30)。脂连蛋白敲除小鼠显现ad样症状(34)。
[0150]
本发明中显示:在ad患者的csf中脂连蛋白浓度降低(图10)。该结果与上次的报道一致(30)。考虑到csf中的蛋白浓度与脑的间质液中的浓度相关(36),这些结果暗示:在ad脑的间质液中脂连蛋白浓度降低,有可能无法维持clsp活性。作为实际的数据,ad患者中的csf脂连蛋白的平均
±
sem浓度为0.31
±
0.13nm,而非ad病例中的该值为0.96
±
0.19nm (非配对t检验、p=0.0065) (图10和表1)。如图5和7所示,推测完全保持clsp活性所需的神经周围局部的脂连蛋白的最低浓度为0.20~0.25nm,这与已降低的ad患者的平均csf脂连蛋白浓度相近。虽然在非ad的神经周围局部存在足够的脂连蛋白量,但暗示了在ad中该位点的脂连蛋白量有可能不足够。作为支持该想法的进一步的数据,发现在神经元内作为人体肽/clsp信号的主要介质的sh3bp5水平在ad皮质中降低(图11)。作为类似的数据,通过先前研究已经显示:作为被人体肽/clsp信号活化的stat3的活性型的、具有磷酸化的第705位的酪氨酸的stat3的水平在ad患者的海马神经元中降低(35)。需要说明的是,这显示了人体肽和clsp与hthnr结合,进而活化由stat3(6)和sh3bp5(24)介导的神经元内信号传递,从而作为
ad保护因子起作用。
[0151]
在先前的研究中,也给出了脂连蛋白在ad患者的血清中上升(29、30)、而在ad脑内降低的数据(图10、以及表1和表2) (30)。该所见的解释之一为以下的想法:脂连蛋白水平因中枢神经系统中发生的一个或几个ad相关异常而降低,然后为了恢复其缺乏,脂肪组织中的脂连蛋白的产生二次上升,其结果在血清中上升。实际上,在先前的研究中暗示:由于脂连蛋白被固定在包含高磷酸化tau的神经内神经原纤维的凝集体中,所以在ad患者的中枢神经系统中脂连蛋白有可能降低(30)。如果该想法正确,则aβ的上升和作为其下游目标的过度磷酸化tau的蓄积成为ad中的脂连蛋白水平降低的主要原因。另一方面,还认为中枢神经系统中的脂连蛋白水平降低是由ad中脂连蛋白的血脑屏障输送受损引起的。目前为止,展示了许多显示在ad的脑内血脑屏障的功能受损的证据(37),但由于尚不存在关于脂连蛋白的血脑屏障输送的数据,所以目前还不明确哪一种想法是正确的。
[0152]
apoe4是主要的ad发病危险因子。迄今为止,通过大量的研究,由apoe4引起的ad发病增加的机制被广泛研究。认为apoe4在aβ依赖性和非依赖性的两种样式中通过多种功能获得和功能丧失机制发挥神经毒性(38)。其中,以下的研究为人所熟知:aβ的产生、aβ从中枢神经的清除、和aβ的凝集形成受到由apoe受体介导的细胞内信息的影响,在apoe4保持者中这些现象倾向于ad发病。
[0153]
本发明中显示:apoe4是比apoe3稍强的clsp的阻碍物质(图2b和c)。与clsp的浓度相比,考虑到cns中的apoe的浓度非常高,轻微的不同没有意义,apoe3和apoe4很有可能同样地使clsp活性降低。然而,若在只有未脂质化(或没有被动员到高密度脂蛋白样脂质粒子中)的游离型apoe可抑制clsp的前提下考虑,则在假设未脂质化的apoe的浓度与clsp的浓度为相同程度的水平的情况下,在脂连蛋白的水平降低的状态(ad患者)下,apoe的clsp阻碍效果的轻微不同可成为左右发病的决定性因素。这种情况下,由于apoe4更强地抑制clsp活性(图2b和c),所以apoe4基因持有者较非apoe4持有者更易受到ad损伤。遗憾的是,目前为止还未找到为了研究该想法的妥当性而应该进行的、定量非脂质化apoe的量的具体方法。
[0154]
在本发明中,根据在ad患者的csf中脂连蛋白水平降低、而且其水平接近于可保护clsp的极限水平(0.3nm)的见解(图10以及表1和表2)、以及sh3bp5和活化的stat3的水平在ad脑内降低(图11) (35)的见解,推测神经元内clsp信号传递在ad脑内下降。
[0155]
然而,在本发明中,测定ad脑神经细胞周边的间质液中的脂连蛋白浓度在技术上几乎是不可能的,所以测定浓度相近的csf中的浓度,据此研究间质液内的现象。
[0156]
而且,在本发明中,直接显示神经元内clsp诱导性信号在ad脑内降低在技术上是不可能的,因此间接地显示。sh3bp5和stat3通过由以各种细胞因子为核心的生理活性物质引起的信号传递路径同时调节,因此可引起sh3bp5水平的降低和stat3的失活,而不会减少clsp诱导信号传递。因此,无法断定clsp诱导性信号的降低是由两者的降低引起的。然而,通常是在ad的cns中发生炎症,其结果产生各种炎性细胞因子,这为人所熟知。而且,上升的各种炎性细胞因子的作用是提高神经元内的活化的stat3和sh3bp5 (stat3的下游目标)的水平。因此,在ad的神经元内活化的stat3和sh3bp5水平低于正常的情况下,考虑存在由释放到周围的各种炎性细胞因子引起的上升机制,认为活化的stat3和sh3bp5的降低显示clsp诱导性信号降低的想法是妥当的。
[0157]
[clspcol]clspcol不受clsp阻碍剂的抑制,具有强效的ad保护活性(图l5)。而且,脂连蛋白的胶原同源区(col)保持着增强和保护内源性野生型clsp的活性。而且,clspcol有效地穿透血脑屏障(图l6)。因此,clspcol等本发明的融合蛋白没有目前所明确的弱点,可作为可通过外周路径送达的ad药候选。
[0158]
然而,clspcol比wt-clspcol和clsp更有效地透过血脑屏障的机制尚未充分阐明。由于clspcol与wt-clspcol之间在效率上存在明显的不同(图l6和表l1),所以clsp的c末端结构域(氨基酸62-146)的缺失很可能会促进效率。即,clsp的c末端侧的一半可包含阻碍血脑屏障转移的区域。另外,通过脂连蛋白的胶原同源区的附着,也有可能使效率提高。
[0159]
clspcol仅被阻碍物质中的钙网蛋白轻度阻碍(图x1、x2)。其具体机制尚不明确,但推测或许是包含人工制作的融合部分的区域对钙网蛋白产生亲和性。然而,预测其阻碍效果弱、而且钙网蛋白的中枢神经系统内浓度低于显示阻碍效果的浓度(小于1nm),所以认为在实际的临床应用上没有形成障碍。
[0160]
上述实施例中使用的材料和方法如下。尚需说明的是,在没有特别记载的情况下,利用本领域技术人员已知的适当的方法/手段实施。
[0161]
[基因和载体]将人clsp插入pcdna3.1-mychis (invitrogen、carlsbad、ca)中,使c末端用mychis标记的人clsp-mychis (clsp-mychis)在哺乳动物细胞中表达(5)。将人载脂蛋白e3、e4、脂连蛋白、膜联蛋白ii和膜联蛋白v的cdna插入到在c末端具有血凝素a(ha)标签的cmv启动子驱动表达载体即pha载体中。插入到pcdna3.1/mychis载体中的小鼠v642i-app cdna记载于现有文献(5)中。将载脂蛋白e3、e4和脂连蛋白cdna插入到pflag载体中,用作在c末端带有flag标签的蛋白表达载体。
[0162]
如文献所记载(5),使用pgex载体(promega、madison、wi)在细菌中生成带有日本住血吸虫谷胱甘肽s-转移酶(gst)标签的重组蛋白。在c末端hibit标签clsp的生成中,将编码hibit氨基酸序列(vsgwrlfkkis)的寡核苷酸、有义引物(seq id no: 4):(5
’‑
cccggggtgagcggctggcggctgttcaagaagattagctgagaattc-3’)、以及反义引物(seq id no: 5):(5
’‑
cccggggtgagcggctggcggctgttcaagaagattagctgagaattc-3’)在体外退火,插入到pgex-2t-clsp质粒的smai-ecori位点。
[0163]
pcmv-sport6载体中的全长人脂连蛋白cdna从invitrogen购入(目录号:6192794、ca)。为了制作c末端用mychisg标记的重组的n末端被gst标记的蛋白,使用利用了以下的诱变引物的kod-plus诱变试剂盒(smk-101东京、日本东洋纺、目录号),使pgex2t-mychis载体的序列突变,产生在c末端添加有甘氨酸残基。
[0164]
有义引物(seq id no: 6):(5
’‑
ggttgagaattcatcgtgactgactgacgatctgcctcgcgcg-3’)、以及反义引物(seq id no: 7):(5
’‑
atgatgatgatgatgatgatcctcttctgagatgagtttttg-3’)。
[0165]
通过kod dna聚合酶(kod-101、东京、日本toyobo、目录号)扩增人脂连蛋白的胶原同源区的cdna。
[0166]
有义引物(seq id no: 8):(5
’‑
ggatccatgagaggatcgcatcaccatcaccatcacgggtcc-3’)、以及反义引物(seq id no: 9):(5
’‑
gaattctcaaggttctcctttcctgccttggattcccggaaagc-3’)。
[0167]
将所扩增的cdna在bamhi-ecori位点亚克隆到pqe30载体中(qiagen、东京、日本)。
[0168]
通过la taq聚合酶(takara公司、目录号rr002a、东京、日本)扩增在n末端具有mychisg标签的脂连蛋白的胶原同源区的cdna。
[0169]
有义引物(seq id no: 10):(5
’‑
aagcttgaacaaaaactcatctcagaagaggatcatcatcatcatcatcatggtatggggcatccgggccataatggggccccaggcc-3’)、以及反义引物(seq id no: 11):(5
’‑
gaattctcaaggttctcctttcctgccttggattcccggaaagcc-3’)。
[0170]
将所扩增的cdna亚克隆到pgex-2t-clsp和-clsp (1-61)质粒中,分别得到了由clsp-mychisg-脂连蛋白的胶原同源区和由clsp (1-61)-mychisg-脂连蛋白的胶原同源区构成的wt-clspcol和clspcol。
[0171]
[重组蛋白]使c末端用mychis标记的gst-人clsp (gst-clsp-mychis)在1mm的异丙基-硫代-β-d-吡喃半乳糖苷中、于37℃下在大肠杆菌bl-21中表达6小时。使gst-clsp-mychis与谷胱甘肽sepharose (ge healthcare)结合,如文献(14)所示,通过在含有1单位/ml的凝血酶的pbs中一同培养,使clsp-mychis部分在25℃下从谷胱甘肽sepharose游离一夜(目录号:t6634-100un、sigma-aldrich、st. lois、mo)。按照相同的方法制作c末端用mychis标记的重组clsp缺失突变体(5)和gst-clsp-hibit。同样地还由在细菌中产生的gst-膜联蛋白ii、膜联蛋白v和sh3bp5制作重组膜联蛋白ii、v和sh3bp5蛋白。使重组gst-mychis和gst-人14-3-3σ在1mm异丙基-硫代-β-d-吡喃半乳糖苷中、于37℃下在大肠杆菌bl-21中表达6小时,与谷胱甘肽sepharose结合,通过在50mm谷胱甘肽的存在下共培养,从谷胱甘肽sepharose中游离,在pbs中进行透析。使脂连蛋白的n末端带有6
×
hisg标签的胶原同源区在1mm异丙基-硫代-β-d-吡喃半乳糖苷中、于37℃下在大肠杆菌m15 [prep4] (qiagen)中表达4小时,与talon metal树脂(clontech、paloalto、ca、usa)结合,按照生产商的指示进行纯化。洗脱的重组6
×
his蛋白通过zeba脱盐柱(pierce)进行脱盐,然后将1/10容量的10
×
pbs添加到脱盐蛋白溶液中。
[0172]
重组人载脂蛋白e3和载脂蛋白e4从peprotech (rocky hill、nj)购入(目录号:350-02和350-04)。人脂连蛋白和三聚体脂连蛋白从biovendor (czeck republic)购入(目录号:rd172029100和rd172023100)。
[0173]
[细胞死亡测定]关于ad的神经细胞死亡测定最初由yamatsuji等人进行(39)。使sh-sy5y细胞在含有10�s的dmem/hamf12混合物(dmem/f12)中增殖。将sh-sy5y细胞以2
×
105/孔在6孔板上接种12~16小时,在血清的不存在下用所指示的载体转染3小时,接下来,在包含/不含clsp和/或clsp修饰物质(与clsp作用的物质)的dmem/f12-10�s中进行培养。转染的24小时后,将培养基交换为包含/不含clsp和/或clsp修饰物质的、含有n2补充剂(invitrogen、
carlsbad、ca)的dmem/f12。在转染开始的48小时后回收细胞,进行使用wst-8细胞死亡测定试剂盒(同仁堂、熊本、日本)的细胞存活(率)测定或使用钙黄绿素am (同仁堂、熊本、日本)的染色、以及锥虫蓝排除细胞死亡测定。sh-sy5y细胞中的转染效率为约80%。所有的细胞死亡实验均以n=3来进行。
[0174]
[抗体]针对与匙孔血蓝蛋白形成复合体的人clsp的n末端肽16氨基酸的肽,产生兔多克隆抗体,用免疫肽进行亲和纯化(hclsp-n抗体)。抗gst-clsp-mychis的兔多克隆抗体通过用细菌中产生的重组gst-clsp-mychis (gst-clsp抗体) (5)免疫而生成。再从使用了重组clsp-mychis的粗血清中亲和纯化抗体。使用14-3-3σ进行亲和纯化。sigma-c抗体通过用人14-3-3σ的c-末端16氨基酸肽对兔进行免疫而产生,再进行亲和纯化。在兔中产生抗sh3bp5的多克隆抗体(命名为“sh3bp5抗体”),然后使用gst-14-3-3σ和gst-sh3bp5进行亲和纯化,再使用gst-sh3bp5进行亲和纯化。
[0175]
针对本发明中使用的肽和蛋白的现成抗体从以下的公司购入:结合有辣根过氧化物酶的flag表位(克隆m2、目录号158592-1mg)、sigma-aldrich;app (22c11、目录号mab348 (注册商标))、chemicon (temecula、ca);myc表位(目录号r950-25)、invitrogen (carlsbad、ca);结合有过氧化物酶的ha (血凝素a)表位(克隆3f10、目录号2013819)、roche diagnostics (alameda、ca);sh3bp5抗体(sab;目录号sc-135617)、biotechnology (santa cruz、ca);sh3bp5单克隆抗体(克隆1d5、目录号h00009467-m02)、abnoba、(台北、中国台湾);hisg单克隆抗体(目录号r940-25)、invitrogen (carlsbad、ca)。
[0176]
[免疫印迹分析]将细胞用pbs洗涤2次,悬浮在50mm的hepes (ph7.4)、150mm的nacl、0.1%的np-40和蛋白酶抑制剂混合物complete (roche diagnostics、alameda、ca)中。在2次冷冻融解后,将细胞裂解物在4℃下以15,000rpm离心分离10分钟。对上清和pull-down沉淀物进行基于标准的或tris-tricine sds聚丙烯酰胺凝胶电泳(sds-page)的分析和免疫印迹分析。在每个泳道将10μg的细胞裂解物直接用于免疫印迹分析(5)。通过为了检测外源表达的v642i-app而使用app抗体的免疫印迹分析同时检测到具有各种长度的内源性野生型app。应留意,由于未知的原因,具有各种长度的内源性野生型app的检测在不同的实验之间是不均匀的。
[0177]
[pull-down分析]重组蛋白与溴化氰活化sepharose 4b的结合按照生产商(amersham pharmacia biotech、uppsala,sweden)的说明书来进行。简单说明如下:使5mg的重组蛋白在偶联缓冲液(含有0.5m nacl的0.1m的nahco3、ph8.3)中与3ml的溴化氰活化sepharose 4b一同旋转,同时在4℃下培养一夜。接下来,将结合有重组蛋白的sepharose在封闭缓冲液(0.2m的甘氨酸、ph8.0)中、于室温下培养2小时,排除非特异性结合。封闭后,将sepharose用偶联缓冲液和含有0.5m nacl的0.1m的乙酸钠缓冲液(ph4)进行洗涤。将结合sepharose 4b在4℃下保存在偶联缓冲液中。
[0178]
将来自过度表达各种蛋白的细胞的裂解物与结合有gst-mychis或clsp-mychis的sepharose 4b在4℃下混合一夜,然后彻底洗涤。然后,对pull-down的沉淀物和细胞裂解物进行sds-page和免疫印迹分析或银染色(和光纯药、东京、日本),研究clsp与蛋白之间的结
合。
[0179]
在实验中,使在细菌中产生c末端用mychis标记的重组clsp、其缺失突变体(δn1、δn2、δc1和ehr)之一并纯化。在4℃下将它们与来自f11细胞的裂解物混合一夜,该裂解物包含c末端用flag标记的载脂蛋白e4或脂连蛋白,然后彻底洗涤。然后将洗涤后的pull-down沉淀物和细胞裂解物进行sds-page展开,进行免疫印迹分析。
[0180]
[在腹腔内注射重组蛋白后调制作为含间质液的脑样品的来自小鼠的脑裂解物]所有的实验步骤均由东京医科大学的动物实验地域委员会批准。对从oriental yeast公司(东京、日本)购入的雌性icr小鼠(8周龄)腹腔内注射作为阴性对照的10nmol的gst-mychisg蛋白、clspcol或wt-clspcol的pbs溶液。注射的1小时后将小鼠用二乙醚(和光纯药、东京、日本)进行麻醉。之后,从心脏吸引血液,在4℃下以4000
×
g离心分离10分钟。使用加入了冰的20ml乳酸林格氏液(大冢制药、东京、日本),通过心脏的左心室灌流脑的血管空间以去除血液。接下来,将小鼠断颈并取出脑。为了洗涤csf的污染,用乳酸林格氏液洗涤全脑一次。然后,在2倍重量的食盐水的存在下进行均质化。将该裂解物在4℃下以4000
×
g离心分离10分钟后,回收上清作为含间质液的脑样品(36)。
[0181]
[人脑脊液和颞叶样品]来自ad患者和对照的死后csf和颞叶样品从duke大学医疗中心、神经内科的kathleen price bryan脑库获得(表1和3)。老人斑和神经炎性斑的病理学分期在“ad联合注册表”(cerad)分期系统(40)下进行,而神经原纤维变化(缠结)的病理学分期在braak分期系统(41)下进行。根据cerad分期视为可能的ad的病例均作为ad病例来计数。该研究由duke大学医疗中心的kathleen price bryan脑库和东京医科大学的伦理委员会批准。
[0182]
[解离常数的测定]使用nano-glo hibit细胞外检测系统(promega、目录号:n2420),按照说明书测定关于载脂蛋白e4 (或脂连蛋白)与clsp之间的结合的解离常数。为了编码重组载脂蛋白e4或脂连蛋白,将含有20pm载脂蛋白e4或脂连蛋白的100μl 50mm的碳酸缓冲液(ph9.6)在96孔板的孔中、于4℃下培养一夜(黑色荧光用板h 目录号:ms-8596kz、住友bakelite、东京、日本)。将蛋白包被板用200μl的pbs洗涤3次。接下来,在各孔中加入包含1%脱脂牛奶的150μl的pbs (gibco)。无需振荡,将它们在室温下培养1小时。将板用200μl的pbs洗涤3次后,在各孔中加入浓度100μl的clsp-hibit的pbs溶液。无需振荡,再将板在4℃下培养一夜,然后用含有0.1%np-40的pbs洗涤5次,之后添加100μl的pbs。之后,将试剂盒中的hibit用底物添加至各孔中。使用wallac arvo
tm x 5 (perkin elmer)测定各孔的所得的化学发光。通过测定用含有浓度阶段性增加的clsp-hibit的100μl pbs填满的孔的化学发光,同时参照制作的标准曲线,推测各孔的clsp-hibit浓度。该实验以n=2来进行。
[0183]
[elisa]从积水medical株式会社(目录号376405、东京、日本)购入现成的人脂连蛋白elisa试剂盒,按照生产商的指示用于测定csf脂连蛋白浓度。在14-3-3σ elisa和sh3bp5的情况下,将含有0.6μg/ml的gst sigma抗体或1μg/ml的sh3bp5单克隆抗体(克隆1d5、目录号h00009467-m02、anoba、台北、中国台湾))的100μl 50mm的碳酸缓冲液(ph9.6)在96孔板(elisa板h、目录号ms-8896fz、住友bakelite、东京、日本)中、于4℃下培养一夜。将该捕捉抗体包被板在各孔中用400μl的洗涤缓冲液(含有0.1%np40的pbs)洗涤3次,然后,无需振
荡,在室温下填满300μl的pvdf封闭试剂(toyobo目录号nypbr01、东京、日本) 1小时。用300μl洗涤缓冲液洗涤3次后,用100μl浓度阶段性增加的重组14-3-3σ或sh3bp5的pbs溶液(用于测定标准曲线)填满板。将人csf样品或人颞叶的裂解物边以250rpm振荡边在室温下培养2小时。之后,用300μl洗涤缓冲液洗涤板。作为检测抗体,使用ab-10快速过氧化物酶标记试剂盒(同仁堂、目录号lk33、熊本、日本)或过氧化物酶标记试剂盒-hn
2 (同仁堂、目录号lk11、熊本)分别调制了过氧化物酶标记sigma-c抗体或sh3bp5抗体。在各孔中添加100μl 1.0μg/ml的检测抗体的can get signal溶液2 (toyobo目录号nkb-301),边以250rpm振荡板边在室温下培养1小时。用300μl洗涤缓冲液洗涤5次后,在孔中添加r&d tmb底物溶液(r&d systems、目录号:dy999),然后将板在室温下培养10分钟。通过添加50μl的h2so4使反应终止。利用wallac arvo
tm x5 (perkin elmer)测定450nm的吸光度。
[0184]
按照生产商的说明书,使用生物素标记试剂盒-nh
2 (同仁堂、目录号:lk03、熊本、日本)调制生物素标记抗hisg抗体。在clspcol和wt-clspcol (顺次包含myc和hisg标签作为结合肽)的elisa的情况下,将包含25μg/ml的clsp-n抗体(捕捉抗体)的100μl 50mm的碳酸缓冲液(ph9.6)在96孔板(elisa板h、目录号ms-8896fz、住友bakelite、东京、日本)中、于4℃下培养一夜。将捕捉抗体包被板用各孔400μl的洗涤缓冲液(含有0.1%tween20的pbs)洗涤3次,然后用300μl的pvdf封闭试剂(toyobo目录号nypbr01、东京、日本)填满,无需振荡,在室温下保持1小时。用300μl洗涤缓冲液洗涤3次后,用包含浓度阶段性增加的gst-mychis、clspcol和wt-clspcol的100μl pbs (用于测定标准曲线)或小鼠脑裂解物填满板,在室温下培养2小时。用300μl洗涤缓冲液洗涤后,用100μl的1000倍稀释的生物素结合抗hisg抗体的can get signal溶液1 (toyobo目录号:nkb-201)填满板,进行培养。在室温下培养1小时。接下来,用300μl洗涤缓冲液洗涤板3次。接下来,用100μl的2000倍稀释的链霉亲和素结合hrp (invitrogen)的can get signal溶液2 (toyobo目录号:nkb-301)填满板,在室温下培养1小时。用300μl洗涤缓冲液洗涤5次后,在板上填充100μl的r&d tm b底物溶液(r&d systems、目录号:dy999),在室温下培养3分钟。通过添加50μl的h2so4使反应终止。利用wallac arvotm x5 (perkin elmer)测定450nm的吸光度。
[0185]
[人样品的免疫组织化学分析]该研究由东京医科大学的伦理委员会批准。由各患者的家属得到知情同意后,按照所建立的步骤在群马老人医学研究病院得到组织学脑样品。按照临床标准患者被诊断为ad,诊断通过尸检下的神经病理学分析得到确认。尸检时,将脑用4%多聚甲醛的pbs溶液(ph7.4)固定,包埋在石蜡中,然后进行神经病理学检查。该研究中使用的大脑皮质和海马是由6名散发性(偶发性)ad患者和5名代表性的运动神经元特异性神经变性疾病即散发性肌萎缩性侧索硬化症(als)患者的样品得到的。
[0186]
对切取的切片进行脱石蜡处理,与pbs进行再水合,然后在抗原解掩蔽液(antigen unmasking solution) (加利福尼亚州burlingame的vector laboratories)中用15分钟去掩蔽。然后,在室温下将切片在包含山羊正常血清和0.3%triton x-100的tbs的封闭溶液中培养20分钟,然后,在含有1%bsa的pbs中、于4℃下与作为阴性对照的5μg/ml的小鼠igg1 (r&d systems目录号mab002、明尼苏达州minneapolis)或sh3bp5 (sab)单克隆抗体克隆pl-a23 (santa cruz biotechnology、目录号sc-135617、santa cruz、加利福尼亚)一同培养3夜。免疫反应性使用tsa (酪酰胺信号放大,tyramide signal amplification)-增强型
荧光素系统(perkin-elmer、waltham、ma) (酪酰胺-红法,tyramide-red 法)进行可视化。在荧光显微镜(biozero、keyence、大阪、日本)下观察荧光标记样品。荧光图像通过nih image j1. 37v进行分析。
[0187]
[神经元中的sh3bp5免疫荧光强度的定量]使用nih image 1.37v定量sh3bp5免疫荧光强度和所选择的神经元的面积。计算每1μm2的神经元的平均sh3bp5免疫荧光强度(a)。同时还定量神经元周围的每1μm2的神经毡的平均免疫荧光强度作为背景免疫荧光(b)。被减掉的平均免疫荧光强度(a-b)用作神经元的平均sh3bp5免疫荧光强度。然后,用神经元面积乘以a-b值,推测神经元中的sh3bp5表达的水平。随机地选择10个神经元,针对各样品计算每个样品的10个神经元中的平均免疫荧光强度。
[0188]
[统计分析]所有数据均利用mac osx软件用的prism7 (graphpad、sandiego、usa)进行分析。细胞死亡实验中的数据用平均值
±
标准偏差表示。其他的所有数据均用平均值
±
sem表示。在由组织学实验和elisa实验得到的数据的分析中采用非配对t检验(两侧)。
[0189]
[表1]已测定csf脂连蛋白浓度的34名尸检受试者的数据分析pmd:到被尸检为止的死后时间;*在将年龄前的“大于(》)”看作“等于”的情况下,p《0.0001。参照表s1的“年龄”。
[0190]
[表2]从表1选出的由年龄81~88岁构成的11个病例的数据分析pmd:到被尸检为止的死后时间。
[0191]
[表3]已测定颞叶或枕叶的外锥体层的神经元内sh3bp5水平的13名尸检受试者的数据
分析[表4]已测定颞叶的细胞裂解物中的sh3bp5水平的20名尸检受试者的数据分析pmd:到被尸检为止的死后时间;*在将年龄前的“大于(》)”看作“等于”的情况下,p《0.876。参照表s3的“年龄”。
[0192]
由于p值小于0.05 (0.032),所以为了进行年龄分析,采用伴有welch修正的非配对t检验。
[0193]
[表5][表s1-1]已检查csf脂连蛋白水平的尸检例的个别数据年龄性别apoepmdcerad阶段或诊断b&b阶段81m347.2正常cerad1bi90m337.4正常cerad1aii88m2317.3正常cerad1ai86m346.1正常cerad1aiii90m334.0正常cerad1bii86m3316.3正常cerad1ai》90m347.7正常cerad1bi》90m3322.3正常cerad1aiii90m233.7正常cerad1aiii》90f235.0正常cerad1bii72f3330.0正常cerad1bii》90f235.2正常cerad1biii85f3313.0正常cerad1aii80f3316正常cerad1aiii
73m339.4可能的adiii79m246.5可能的adiii[表6][表s1-2]已检查csf脂连蛋白水平的尸检例的个别数据年龄性别apoepmdcerad阶段或诊断b&b阶段82m341.3adv83m332.0adv73m3422.3adv76m347.0adv80m446.5adv71m346.5adv78m348.0adv85m3423.5adv75m3412.2adv77m3310.7adv83m3312.8adv79f4416.2adv80f3316adv79f448.0adv77f4435.4adv81f4414.7adv84f345.9adv75f348.8advapoe:2个载脂蛋白e等位基因名称用数字表示;pmd:到被尸检为止的死后时间、b&b阶段:braak&braak阶段;2个可能的ad病例被计为ad病例。
[0194]
所有ad病例和非ad病例的平均
±
sem年龄分别大于78.5
±
0.9岁和86.3
±
1.4岁(非配对t检验、在将年龄前的“大于”看作“等于”的情况下p《0.0001)。所有的ad病例和非ad病例的平均
±
sem pmd分别为11.7
±
1.8小时和11.5
±
2.1小时(非配对t检验、p=0.96)。
[0195]
[表7][表s2]已测定颞叶或枕叶的外锥体层的神经元内sh3bp5水平的尸检例的个别数据
颞叶或枕叶的外锥体层的切片是由被尸检的ad和als患者得到的。cdr:临床痴呆评价、ne:没有检查。
[0196]
所有als患者和ad患者的平均
±
sem年龄分别为66.7
±
2.8和75.9
±
5.1岁(非配对t检验、p=0.158)。
[0197]
[表8][表s3]已测定颞叶的细胞裂解物中的sh3bp5水平的尸检例的个别数据apoe:2个载脂蛋白e等位基因名称用数字表示;pmd:到被尸检为止的死后时间、b&b阶段:braak&braak阶段。
[0198]
所有ad病例和非ad病例的平均
±
sem年龄分别大于79.9
±
2.9岁和79.4
±
1.3岁(非配对t检验、在将年龄前的“大于”看作“等于”的情况下p《0.876)。所有ad病例和非ad病
例的平均
±
sem pmd分别为12.7
±
3.0小时和16.7
±
3.1小时(非配对t检验、p=0.368)。
[0199]
[表9][表l1]含间质液的脑裂解物和血清中的clspcol和wt-clspcol的elisa数据通过测定浓度阶段性增加的重组蛋白(n=3),制作了标准的剂量反应曲线。在含isf的脑裂解液和血清中的clspcol或wt-clspcol浓度的测定中,对包含10μl含间质液的脑裂解液的90μl pbs (
×
10 isf裂解液)或包含2μl血清的98μl pbs (血清
×
50)进行elisa (n=3)。关于浓度阶段性增加的标准重组蛋白(gst-mychisg、wt-clspcol和clspcol;浓度0.15~10nm)、
×
10 isf裂解物以及
×
50血清的实测数值见abs 450列。接下来,计算3个数
matsuoka,y.ahumaninderivativereducesamyloidbetaaccumulationandamelioratesmemorydeficitintripletransgenicmice.plosone6:e16259(2011).26.zhang,w.etal.s14g-humaninimprovescognitivedeficitsandreducesamyloidpathologyinthemiddle-agedappswe/ps1de9mice.pharmacol.biochem.behavior100,361-369.(2012).27.yin,r.etal.protectiveeffectsofcolivelinagainstalzheimer’sdiseaseinapdappmousemodel.cell.physiol.biochem.38,1138-1146(2016).28.combst.p.,berga.h.,obicis.,schererp.e.,&rossettil.endogenousglucoseproductionisinhibitedbytheadipose-derivedproteinacrp30.j.clin.investig.108,1875-1881(2001).29.une,k.etal.adiponectininplasmaandcerebrospinalfluidinmciandalzheimer’sdisease.eur.j.neurol.18,1006-1009(2011).30.waragai,m.etal.possibleinvolvementofadiponectin,theanti-diabetesmolecule,inthepathogenesisofalzheimer’sdisease.j.alzheimersdis.52,1453-9(2016).31.ng,r.c.,&chan,k.h.potentialneuroprotectiveeffectsofadiponectininalzheimer’sdisease.int.j.mol.sci.18,pii:e592(2017).32.himbergen,t.m.v.etal.biomarkersforinsulinresistanceandinflammationandtheriskforall-causedementiaandalzheimerdiseaseresultsfromtheframinghamheartstudy.arch.neurol.69,564-600(2012).33.garcia-casaresn.etal.alzheimer’slikebrainchangescorrelatewithlowadiponectinplasmalevelsintype2diabeticpatients.j.diabetescomplicat.30,281-286(2016).34.ng,r.c.l.etal.chronicadiponectindeficiencyleadstoalzheimer’sdisease-likecognitiveimpairmentsthroughampkinactivationandcerebralinsulinresistanceinagedmice.mol.neurodegener.11,71(2016).35.chiba,t.etal.amyloid-betacausesmemoryimpairmentbydisturbingthejak2/stat3axisinhippocampalneurons.mol.psychiatry14,206-222(2009).36.liu,x.etal.unbounddrugconcentrationinbrainhomogenateandcerebralspinalfluidatsteadystateasasurrogateforunboundconcentrationinbraininterstitialfluid.drugmetab.dispos.37,787-93(2009).37.sweeney,m.d.,sagare,a.p.,&zlokovic,b.v.blood-brainbarrierbreakdowninalzheimerdiseaseandotherneurodegenerativedisorders.nat.rev.neurol.14,133-150(2018).38.liu,c.-c.,kanekiyo,t.,xu,h.,&bu,g.apolipoproteineand
alzheimer disease: risk, mechanisms, and therapy nat. rev. neurol. 9, 106-118 (2012).39. yamatsuji, t., et al. g protein-mediated neuronal dna fragmentation induced by familial alzheimer’s disease-associated mutants of app. science 272, 1349-1352 (1996).40. mirra, s.s.et al. the consortium to establish a registry for alzheimer’s disease (cerad). part ii. standardization of the neuropathologic assessment of alzheimer’s disease. neurology 41, 479-486 (1999).41. braak h., & braak e. neuropathological staging of alzheimer-related changes. acta neuropathol (berl) 82, 239-59 (1991).42. pajvani, u. b. et al. structure-function studies of the adipocyte-secreted hormone acrp30/adiponectin. implications for metabolic regulation and bioactivity j. biol. chem. 278, 9073 (2003)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
