1.本发明属于生物降解聚合物材料领域,具体涉及一种低环状副产物的聚丁二酸丁二醇酯的制备方法。
背景技术:
2.聚丁二酸丁二醇酯(pbs)及其共聚酯是一类具有良好生物可降解性和综合性能的脂肪族聚酯,通常采用丁二酸和1,4-丁二醇直接酯化缩聚制备,是最有希望替代传统聚烯烃塑料的材料之一。pbs由于优异的生物降解性和力学性能在一次性餐盒、吸管等食品接触材料领域有很强的应用潜力。但众所周知,二元酸、二元醇反应得到的聚酯不可避免的会产生环状副产物,聚酯中的环状副产物(如环状单体、二聚体、三聚体和四聚体)的存在使得pbs不能满足食品接触材料的要求,尤其是环状单体和环状二聚体比相应的更高级的低聚物经受更严重的迁移,但目前对pbs的环状副产物的研究较少。
3.ep-a2623540公开了一种纯化脂肪族聚酯如pbs的方法,其中环状副产物通过溶剂萃取法除掉,但该萃取方法的缺点是会造成聚酯发生降解,分子量不可控,力学性能变差。
4.cn111372972公开了一种在脱气装置中纯化脂肪族聚酯的方法,环状副产物通过夹带剂除掉,夹带剂优选水,该方法同样会造成聚酯降解,且操作复杂。
5.综上所述,亟需一种高效、易于实现工业化的方法制备低环状副产物的聚丁二酸丁二醇酯。
技术实现要素:
6.本发明针对现有技术中存在的上述缺点,提供一种聚丁二酸丁二醇聚酯(pbs)的制备方法。该方法可以在合成过程中有效减少环状副产物的产生,得到的pbs可直接用于食品接触材料,省去pbs后处理步骤。
7.为达到上述发明目的,本发明采用的技术方案如下:
8.一种低环状副产物的聚丁二酸丁二醇酯(pbs)的制备方法,所述方法包含以下步骤:
9.s1:将钛酸酯、丁二酸和1,4-丁二醇加入到反应釜进行酯化反应;
10.s2:反应釜内加入固体负载的锡催化剂和助剂冠醚进行缩聚反应,得到聚合物熔体,经造粒得到聚丁二酸丁二醇酯聚合物。
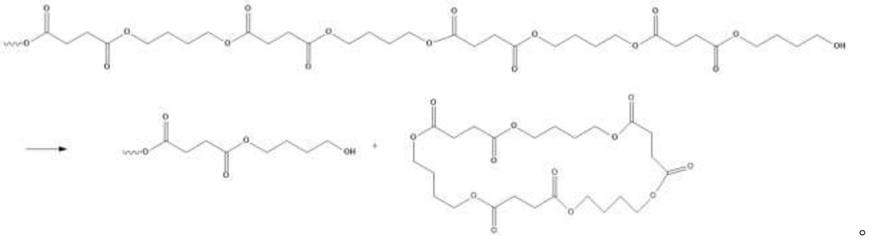
11.在pbs制备领域,二元酸、二元醇反应得到的聚酯不可避免的会产生环状副产物,研究认为主要是由大分子链端进攻酯基裂解而成环形成的,下式为其中一种环状三聚体的产生过程:
[0012][0013]
目前只报道过溶剂萃取、夹带剂脱除pbs中环状副产物的方法,但这两种方法会造成聚酯降解,分子量不可控。本发明人发现可使用开环催化剂如锡化合物将环状副产物开环继续反应生成pbs,但环状副产物含量较少,单纯使用锡化合物进行开环面临底物(环状副产物)少催化效率低等问题,这就需要增加锡化合物使用量,增加了生产成本,通过使用分子筛等具有小分子吸附功能的载体能有效吸附环状副产物,大大增加了环状副产物与催化剂接触效率。且锡化合物本身也具有酯交换活性,通过使用助剂冠醚,冠醚环中氧原子与金属离子络合,大大增强了锡催化剂的开环催化活性,弱化锡催化剂的酯交换催化活性。同样,分子筛等载体也可有效吸附冠醚,使冠醚环和锡化合物有效结合。
[0014]
本发明中,s1所述钛酸酯为ti(or)4,其中r为1~10个碳原子的烷基,优选钛酸四丁酯、钛酸四异丙酯、钛酸四乙酯和钛酸四甲酯中的一种或多种;优选地,所述钛酸酯以元素ti计含量为50~300ppm,以聚酯总质量计。
[0015]
本发明中,s1所述丁二酸和1,4-丁二醇摩尔比为1:1.1~1:1.5,优选1:1.1~1:1.3。
[0016]
本发明中,所述s1在常压180~250℃进行酯化反应,脱出副产物水。
[0017]
本发明中,s1当总酯化率达到95%以上时,结束酯化反应。
[0018]
本发明中,s2所述固体负载的锡催化剂中锡化合物为亚锡类化合物,优选氯化亚锡、辛酸亚锡、苯甲酸亚锡、氧化亚锡、异丁酸亚锡、草酸亚锡中的一种或多种,更优选氯化亚锡、辛酸亚锡、苯甲酸亚锡中的一种或多种,进一步优选氯化亚锡和/或辛酸亚锡,最优选氯化亚锡;优选地,所述锡化合物含量以载体质量计为1~5wt%,优选2~3.5wt%;。
[0019]
本发明中,s2所述固体负载的锡催化剂的载体为分子筛、活性炭、硅藻土、蒙脱土中的一种或多种,优选分子筛,更优选50-200目分子筛;优选地,所述固体负载的锡催化剂加入量为丁二酸质量的0.02~2%,优选0.2~1%。
[0020]
本发明中,s2所述冠醚为分子中至少含有4个-氧-亚甲基-结构单元的大环多醚,优选15-冠-5、18-冠-6、1,10二氮18-冠-6、二苯并18-冠-6、二环己烷并18-冠-6、二苯并18-冠-6中的一种或多种,更优选15-冠-5和/或18-冠-6,最优选18-冠-6;优选地,所述冠醚以固体负载的锡催化剂质量计,使用量为1~5wt%,优选2~4wt%。
[0021]
本发明中,所述s2开始缩聚反应时,先抽真空至绝压1000~30,000pa,持续10~60min,再抽真空至绝压100pa以下的高真空。
[0022]
本发明中,所述s2缩聚反应温度220~260℃,高真空缩聚时间60~200min。
[0023]
本发明中,所述s2缩聚反应后经水冷切粒得到聚丁二酸丁二醇酯聚合物。
[0024]
本发明的另一目的在于提供一种制备所述固体负载的锡催化剂的制备方法。
[0025]
一种制备固体负载的锡催化剂的制备方法,所述制备方法为将锡化合物、载体溶
于溶剂中,经超声、烘干至恒重。
[0026]
本发明中,所述锡催化剂的制备方法中溶剂为醇、酮、醚中的一种或多种,优选c2~c4小分子醇,更优选乙醇;优选地,所述溶剂用量为载体质量的3~10倍,优选5~7倍。
[0027]
本发明中,所述锡催化剂的制备方法中超声时的温度为20~60℃,优选30~50℃,超声时间为3~10h,优选4~6h。
[0028]
本发明中,所述锡催化剂的制备方法中烘干温度为80~150℃,优选100~120℃。
[0029]
本发明的又一目的在于提供一种聚丁二酸丁二醇酯产品。
[0030]
一种聚丁二酸丁二醇酯,采用上述的低环状副产物聚丁二酸丁二醇酯的制备方法制备获得,或采用上述固体负载的锡催化剂的制备方法制备的催化剂催化制备获得,所述聚丁二酸丁二醇酯中环状副产物含量≤1wt%,优选≤0.5wt%,以聚丁二酸丁二醇酯总质量计。所述环状副产物为下式所示化合物,其中n为小于10的正整数,当n为1时为环状单体,n为2时为二聚体、n为3时为三聚体,
[0031][0032]
与现有技术相比,本发明技术方案积极效果在于:
[0033]
(1)在合成过程中即有效控制环状副产物的产生,pbs环状副产物含量≤0.5wt%;
[0034]
(2)减少pbs后处理步骤,有效控制分子量分布,力学性能优良。
具体实施方式
[0035]
下面结合实施例对本发明作进一步的说明,需要说明的是,实施例并不构成对本发明要求保护范围的限制。
[0036]
原料:
[0037]
丁二酸,优等品,山东飞扬化工有限公司;
[0038]
1,4-丁二醇(bdo),工业级,新疆美克化工股份有限公司;
[0039]
钛酸四丁酯98%,试剂级,阿拉丁试剂有限公司;
[0040]
钛酸四异丙酯98%,试剂级,阿拉丁试剂有限公司;
[0041]
氯化亚锡98%,试剂级,阿拉丁试剂有限公司;
[0042]
辛酸亚锡98%,试剂级,阿拉丁试剂有限公司;
[0043]
4a分子筛,50目,阿拉丁试剂有限公司;
[0044]
4a分子筛,100目,阿拉丁试剂有限公司;
[0045]
4a分子筛,200目,阿拉丁试剂有限公司;
[0046]
硅藻土,中位粒径19.6微米,阿拉丁试剂有限公司;
[0047]
乙醇99%,试剂级,阿拉丁试剂有限公司;
[0048]
15-冠-5,98%,试剂级,阿拉丁试剂有限公司;
[0049]
18-冠-6,98%,试剂级,阿拉丁试剂有限公司。
[0050]
除特殊说明外,本发明所用设备和方法均为本领域通用的设备和方法。其中,样品的分子量是采用美国waters公司的1515-2414型凝胶渗透色谱(gpc)仪来测定,其中六氟异丙醇为流动相,流出速度为1ml/min,柱温为30℃,标准样为聚苯乙烯。
[0051]
力学性能通过以下方法测试:拉伸性能采用力学试验机测试(instron 5960),拉伸速度为50mm/min。
[0052]
环状副产物通过赛默飞tsq 8000evo气相色谱-质谱联用(gc-ms)进行表征。将24.41mg的样品溶解在1.2ml的二氯甲烷中中,将安瓿瓶放置在滚轴混匀仪上30分钟,电离通过电子碰撞电离进行。采用单一分辨率。
[0053]
固体负载的锡类催化剂制备:
[0054]
实施例1
[0055]
称量0.5g氯化亚锡、50g 50目4a分子筛,溶于150g乙醇中,20℃超声3小时,80℃烘干直至重量恒定,制得催化剂a。
[0056]
实施例2
[0057]
称量2.5g氯化亚锡、50g 100目4a分子筛,溶于500g乙醇中,60℃超声10小时,150℃烘干直至重量恒定,制得催化剂b。
[0058]
实施例3
[0059]
称量1.5g氯化亚锡、50g 200目4a分子筛,溶于300g乙醇中,40℃超声5小时,110℃烘干直至重量恒定,制得催化剂c。
[0060]
实施例4
[0061]
称量1.5g辛酸亚锡、50g硅藻土,溶于300g乙醇中,40℃超声5小时,110℃烘干直至重量恒定,制得催化剂d。
[0062]
制备聚酯:
[0063]
实施例5
[0064]
在5l聚酯釜中加入丁二酸10mol、丁二醇15mol、3.67g钛酸四丁酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至250℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。加入23.6g催化剂a和1.18g 18-冠-6,反应釜逐渐抽真空至20,000paa,持续30min,然后逐渐抽真空至90paa,升温至220℃并保持,进行缩聚反应,反应200min,得到聚合物熔体,经水冷切粒,得到产品。
[0065]
实施例6
[0066]
在5l聚酯釜中加入丁二酸10mol、丁二醇11mol、0.61g钛酸四丁酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至180℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。加入0.24g催化剂b和0.0024g 18-冠-6,反应釜逐渐抽真空至1000paa,持续20min,然后逐渐抽真空至90paa,升温至260℃并保持,进行缩聚反应,反应100min,得到聚合物熔体,经水冷切粒,得到产品。
[0067]
实施例7
[0068]
在5l聚酯釜中加入丁二酸10mol、丁二醇12mol、1.83g钛酸四丁酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至220℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。加入7.08g催化剂c和0.21g 18-冠-6,反应釜逐渐
抽真空至2000paa,持续50min,然后逐渐抽真空至90paa,升温至240℃并保持,进行缩聚反应,反应150min,得到聚合物熔体,经水冷切粒,得到产品。
[0069]
实施例8
[0070]
在5l聚酯釜中加入丁二酸10mol、丁二醇12mol、1.53g钛酸四异丙酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至220℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。加入7.08g催化剂d和0.21g 15-冠-5,反应釜逐渐抽真空至2000paa,持续50min,然后逐渐抽真空至90paa,升温至240℃并保持,进行缩聚反应,反应150min,得到聚合物熔体,经水冷切粒,得到产品。
[0071]
对比例1
[0072]
与实施例6比较,不同在于酯化后不加入固体负载的锡催化剂和助剂冠醚。
[0073]
在5l聚酯釜中加入丁二酸10mol、丁二醇12mol、1.83g钛酸四丁酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至220℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。反应釜逐渐抽真空至2000paa,持续50min,然后逐渐抽真空至90paa,升温至240℃并保持,进行缩聚反应,反应150min,得到聚合物熔体,经水冷切粒,得到产品。
[0074]
对比例2
[0075]
与实施例6比较,不同在于酯化后只加入固体负载的锡催化剂,不加入助剂冠醚
[0076]
在5l聚酯釜中加入丁二酸10mol、丁二醇12mol、1.83g钛酸四丁酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至220℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。加入7.08g催化剂c,反应釜逐渐抽真空至2000paa,持续50min,然后逐渐抽真空至90paa,升温至240℃并保持,进行缩聚反应,反应150min,得到聚合物熔体,经水冷切粒,得到产品。
[0077]
对比例3
[0078]
与实施例6比较,不同在于酯化后加入锡化合物和助剂冠醚
[0079]
在5l聚酯釜中加入丁二酸10mol、丁二醇12mol、1.83g钛酸四丁酯,釜内保持常压,恒速搅拌,升温至150℃,开始反应,1h内逐渐升温至220℃,待反应釜内馏出副产物水的量达到理论出水量95%时,完成酯化过程。加入7.08g氯化亚锡和0.21g 18-冠-6,反应釜逐渐抽真空至2000paa,持续50min,然后逐渐抽真空至90paa,升温至240℃并保持,进行缩聚反应,反应150min,得到聚合物熔体,经水冷切粒,得到产品。
[0080]
表1 pbs性能
[0081]
组别低聚物含量/%pbs数均分子量pbs重均分子量pdi拉伸强度/mpa实施例40.5101,024364,8653.6139实施例50.898,735360,5103.6536实施例60.2110,982388,9403.5045实施例70.699,153361,5243.6537对比例11.685,464361,9584.2430对比例21.490,317355,5423.9431对比例31.393,841350,1423.7332
[0082]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,
任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
