1.本发明属于药物合成技术领域,具体涉及琥珀酸去甲文拉法辛原料药的制备方法。
背景技术:
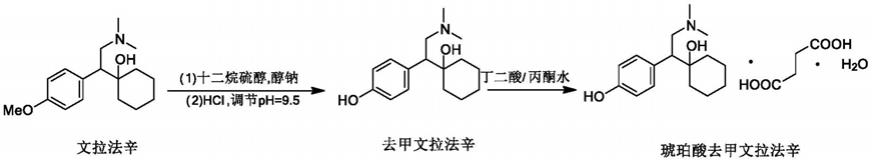
2.琥珀酸去甲文拉法辛是由美国惠氏公司开发的5-羟色胺-去甲肾上腺素再摄取抑制剂类的抗抑郁新药。原研公司发明专利cn1501909a公开了琥珀酸去甲文拉法辛化合物的制备方法,具体为:将文拉法辛(55g),十二烷基硫醇,乙醇钠的乙醇溶液加入到加压容器中,将温度升至150℃,并将此反应混合物搅拌2天,然后降温并过滤此溶液,用氯化氢水溶液将滤液ph调至9.5,抽滤,滤饼用乙醇洗涤真空干燥,得42g游离碱,收率80.43%。另一实施例则采用十二烷基硫醇(122g),文拉法辛和甲醇钠的甲醇溶液,溶剂peg400加热至190℃,蒸馏掉甲醇并在190℃搅拌反应2小时,然后降温加入2-丙醇(450g),并用盐酸水溶液调ph 至9.5,抽滤,滤饼用2-丙醇、甲苯、2-丙醇和水洗涤,得去甲文拉法辛,收率为82.5%。第二步去甲文拉法辛在丙酮和水中成琥珀酸盐一水合物收率最高为85.8%,两步拉通最高收率为70.8%,拉通收率偏低;大生产碱化温度控制在0~10℃,调ph 9~10,离心困难,且存在穿滤,导致去甲文拉法辛滤饼中残留大量的盐,最终导致原料药炽灼残渣高于药典标准不得过0.1%要求,不易产业化。
3.
技术实现要素:
4.本发明目的在于提供一种高收率、可生产放大制备灼烧残渣符合药典要求(不得过0.1%) 的琥珀酸去甲文拉法辛原料药的方法。
5.本发明提供了一种高收率制备琥珀酸去甲文拉法辛的制备方法,所述的制备方法包括如下步骤:
6.步骤1:文拉法辛和十二烷基硫醇钠作反应,制备得去甲文拉法辛反应液;
7.步骤2:将所述反应液用酸化试剂调ph至1.0~4.0,用有机溶剂提取不成盐的杂质;
8.步骤3:将步骤2得到的水相,控温20℃~50℃,用碱化剂调ph 9.0~10.0;
9.步骤4:将步骤3碱化后体系离心,滤饼检测炽灼残渣,若大于0.1%则用水打浆,至离心滤饼炽灼残渣不得过0.1%;
10.步骤5:将步骤4制备得到的去甲文拉法辛湿品直接投料,加入丙酮水溶液和琥珀酸升温溶解后,降温过滤,减压真空干燥得琥珀酸去甲文拉法辛原料药。
11.在本发明的上述实施方案中,步骤1中所述的反应是在n-甲基吡咯烷酮中进行的;可选地,所述n-甲基吡咯烷酮与文拉法辛的比为(2ml~5ml):1g,优选地,为3ml:1g。
12.在上述实施方案中,步骤1中文拉法辛与十二烷基硫醇钠的摩尔比为1:(2~4),优选地,为1:3.5。
13.在上述实施方案中,步骤2中所述的酸化试剂为盐酸。
14.在上述实施方案中,步骤2中所述有机溶剂为乙酸乙酯或二氯甲烷。
15.在上述实施方案中,步骤3中所述碱化剂为氢氧化钠、氢氧化钾、碳酸钾或碳酸钠。
16.在上述实施方案中,步骤3中所述控温为20℃~30℃或40℃~50℃。
17.在上述实施方案中,步骤4中琥珀酸与步骤1中文拉法辛重量比为0.37:1。
18.在上述实施方案中,步骤4中丙酮水溶液中丙酮:水的体积比为2.5:1。
19.在上述实施方案中,步骤5中所述升温溶解的温度为45℃~70℃,优选地,为55℃~65℃。
20.在上述实施方案中,步骤4中所述降温为:快速降温至30℃~40℃,于30~40℃保温搅拌结晶约2h;再降温至5℃搅拌结晶2h。
21.本发明的有益结果在于:
22.本发明提供的琥珀酸去甲文拉法辛的制备方法拉通收率85.0%(从文拉法辛计算),比专利cn1501909a报道两步最高拉通收率70.8%高,同时本发明解决了大生产上如何制备残渣合格的琥珀酸去甲文拉法辛原料药的技术问题。
具体实施方式
23.下列实施例进一步说明本发明,但本发明的保护范围并不受其限制。
24.对比例 琥珀酸去甲文拉法辛的制备
25.于反应釜中加入92.22kg文拉法辛,十二烷基硫醇钠(3.5eq),n-甲基吡咯烷酮(3.0ml/g),搅拌升温至170~180℃,保温搅拌反应6h,降温至50℃以下,加入水(5ml/g)和乙酸乙酯(4ml/g),控温0~10℃用浓盐酸调节ph至1,分液,水相用乙酸乙酯(4ml/g)洗涤,水相控温0~10℃滴加40%氢氧化钠水溶液调节ph至9~10,过滤,产品颗粒较细,离心困难,且穿滤,滤饼粘稠,打浆困难,得去甲文拉法辛湿品,重量91.18(kg),含水13.8kg。
26.反应釜中加入去甲文拉法辛湿品91.18(kg),再依次加入丁二酸(琥珀酸)0.37g/g(以文拉法辛投料量计)、丙酮5.27g/g(根据100%理论收率得到的去甲文拉法辛计算),纯化水(2.61m
‑ꢀ
x)kg,(m为文拉法辛投料量92.22kg,x为去甲文拉法辛湿品重量91.18kg)搅拌加热升温至55~65℃,体系溶清后,热过滤,搅拌加热至55~65℃,体系溶清后,快速降温至30~40℃,于30~40℃保温搅拌结晶约2h。再降温至5℃搅拌结晶2h。过滤,滤饼于45~55℃减压干燥 10h,得类白色至白色结晶性粉末109.7kg,收率为76.9%,炽灼残渣3.8%;
27.残渣不合格返工:上步得到的琥珀酸去甲文拉法辛投料90.14kg加入反应釜,加入3.55g/g 丙酮和1.42g/g纯化水升温55.0~65.0℃后降温至-10.0~5℃,搅拌2.0~3.0h离心,滤饼于 50.0~60.0℃,真空度不低于-0.08mpa减压干燥至水分合格,得80.02kg,收率88.77%,检测炽灼残渣3.5%,标准限度0.1%,不合格;
28.再次仅用水打浆返工:反应釜中加入77.02kg琥珀酸去甲文拉法辛,加入6.0g/g水,20.0~ 30.0℃打浆6小时
±
10分钟,离心后用1.58g/g丙酮洗涤滤饼,滤饼于50.0~
60.0℃,真空度不低于-0.08mpa减压干燥至水分合格,得50.05kg,收率64.96%,炽灼残渣0.02%。
29.实施例1琥珀酸去甲文拉法辛的制备
30.于反应釜中加入89.62kg文拉法辛,十二烷基硫醇钠(3.5eq),n-甲基吡咯烷酮(3.0ml/g),搅拌升温至170~180℃,保温搅拌反应6h,降温至50℃以下,加入水(5ml/g)和乙酸乙酯(4ml/g),控温0~10℃用浓盐酸调节ph至1,分液,水相用乙酸乙酯(4ml/g)洗涤,水相控温20~ 30℃滴加40%氢氧化钠水溶液调节ph至9~10,过滤,滤饼测炽灼残渣2.5%,滤饼用纯化水6.8g/g,20~30℃打浆0.5~1.0小时,离心,滤饼检测炽灼残渣0.06%,得去甲文拉法辛湿品,重量115.33(kg),含水其中,m为文拉法辛投料量89.62kg。
31.反应釜中加入去甲文拉法辛湿品115.33(kg),再依次加入丁二酸(琥珀酸)0.37g/g(以文拉法辛投料量计)、丙酮5.27g/g(根据100%理论收率得到的去甲文拉法辛计算),纯化水 (2.61m-x)g,(m为文拉法辛投料量89.62kg,x为去甲文拉法辛湿品重量115.33)搅拌加热升温至55~65℃,体系溶清后,热过滤,体系快速降温至30~40℃,于30~40℃保温搅拌结晶约2h。再降温至5℃搅拌结晶2h。过滤,滤饼于45~55℃减压干燥10h,得类白色至白色结晶性粉末109.7kg,收率为85.0%,炽灼残渣0.02%(收率从文拉法辛计算)。
32.实施例2琥珀酸去甲文拉法辛的制备
33.于反应釜中加入85.12kg文拉法辛,十二烷基硫醇钠(3.5eq),n-甲基吡咯烷酮(3.0ml/g),搅拌升温至170~180℃,保温搅拌反应6h,降温至50℃以下,加入水(5ml/g)和乙酸乙酯(4ml/g),控温0~10℃用浓盐酸调节ph至4,分液,水相用二氯甲烷(4ml/g)洗涤,水相控温40~50℃滴加40%氢氧化钠水溶液调节ph至9~10,过滤,滤饼测炽灼残渣2.7%,滤饼用纯化水6.8g/g,20~30℃打浆0.5~1.0小时,离心,滤饼检测炽灼残渣0.05%,得去甲文拉法辛湿品,重量108.11(kg),含水其中,m为文拉法辛投料量85.12kg。
34.反应釜中加入去甲文拉法辛湿品108.11(kg),再依次加入丁二酸(琥珀酸)0.37g/g(以文拉法辛投料量计)、丙酮5.27g/g(根据100%理论收率得到的去甲文拉法辛计算),纯化水 (2.61m-x)g,(m为文拉法辛投料量85.12kg,x为去甲文拉法辛湿品重量108.11kg) 搅拌加热升温至55~65℃,体系溶清后,热过滤,快速降温至30~40℃,于30~40℃保温搅拌结晶约2h。再降温至5℃搅拌结晶2h。过滤,滤饼于45~55℃减压干燥10h,得类白色至白色结晶性粉末107.90kg,收率为88.0%,炽灼残渣0.01%(收率从文拉法辛计算)。
35.最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管通过参照本发明的优选实施例已经对本发明进行了描述,但本领域的普通技术人员应当理解,可以在形式上和细节上对其作为各种各样的改变,而不偏离所附权利要求所限定的本发明的精神和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。