ngs肿瘤基因突变检测降噪方法
技术领域
1.本发明涉及ngs肿瘤基因突变检测技术领域,具体为ngs肿瘤基因突变检测降噪方法。
背景技术:
2.高通量基因检测可为肿瘤的临床治疗提供更精准的治疗方案,基因检测结果的准确性至关重要。目前分析肿瘤体细胞突变的常用软件有mutect2、strelka2、varscan等,但这些软件给出的最终结果往往无法直接使用,存在一定的假阳性和假阴性突变。造成假阳性和假阴性的因素较多,主要包括样本局部降解、样本氧化损伤、样本污染、测序仪测序错误、试剂和系统噪音等。
3.一个肿瘤样本通常有几十甚至上百个候选突变位点,其中大部分为假阳性突变。这些候选位点的真假判定,需要生信分析人员掌握非常丰富的经验,而且耗费大量的人力和时间成本。
4.本发明针对以上提到的可能造成假阳性和假阴性突变的众多因素,依次进行降噪过滤和验证,实现自动化和流程化,最终呈现真实有效的突变。
技术实现要素:
5.本发明的目的在于提供ngs肿瘤基因突变检测降噪方法,解决了背景技术中所提出的问题。
6.为实现上述目的,本发明提供如下技术方案:ngs肿瘤基因突变检测降噪方法,包括以下步骤:
7.步骤一:提前准备好四个数据库:靶点数据库、人群频率数据库、噪音数据库、str数据库。靶点数据库的收集主要包括两个方面,一个是从nccn、fda、nmpa等机构发布的文档或者其官方文档中整理,另一个是下载clinvar、oncokb、cosmic数据库中致病或可能致病的突变位点(不包含短串联重复区域的突变),最后编写脚本(python/r等编程语言)将这两部分的突变位点合并,并以hgvs规则进行注释,以ghgvs作为每个突变位点的标签。人群频率数据库的准备,需要下载dbsnp、千人基因组、gnomad、exac等数据库的vcf文件,编写脚本提取人群频率大于千分之一(只要有一个数据库满足条件即可)的突变位点,同样以ghgvs作为每个突变位点的标签。噪音数据库依赖本地已分析的肿瘤样本数据,样本数量越多越好。其中高频出现(大于1%)的非致病突变位点,按照突变丰度的波动范围(突变丰度的最小值到最大值)进行分类,一般胚系突变的波动范围在40%~60%或者90%~100%,试剂和系统噪音突变一般在0~5%或者在某个恒定的值(如某个噪音突变在收录的肿瘤样本中都以20%左右的突变丰度出现)。把这两部分突变整理合并,并同样以ghgvs作为每个突变位点的标签。str数据库,编程提取人类基因组或某个bed区间内str(短串联重复)区域重复单元的插入或缺失突变,同样以ghgvs作为每个突变位点的标签;
8.步骤二:复杂位点合并:这里以一个复杂的egfr 19del突变举例,mutect2等分析
软件分析出chr7:g.55242469_55242477del和chr7:g.55242478g》c这两个突变,这两个突变的丰度都在32%左右,而且通过查看bam文件,这两个突变发生在相同的reads上,所有这两个突变其实是一个复杂突变,需要将其合并,最终合并成chr7:g.55242469_55242478delinsc;
9.步骤三:假阳性过滤:这一步主要包含umi过滤、链偏好性过滤、突变单元分布过滤、非致病位点过滤、噪音过滤五个小步骤。umi过滤:用samtools工具从原始的bam文件中提取每个突变位点上下游各250bp范围的所有reads,对各个reads的umi进行统计,最终统计出每个umi所拥有的reads数(筛选大于等于2个reads支持的umi,短串联重复区域突变的阈值提高到3),及其在突变reads和未突变reads中的比例(过滤同时出现在未突变reads中的umi),筛选后的umi数大于等于3的突变位点可以保留。链偏好性过滤:mutect2等软件一般也有这方面的过滤,只是mutect2的过滤过于严苛,很多时候会过滤掉真实突变。本发明的方法是把mutect2分析结果中的sb值提取出来,再用r语言中的fisher.test函数计算链偏好性p值,过滤p值小于0.05的突变位点。突变单元分布过滤:以突变位点为中心,左右各延伸10bp,把这段长约20bp的片段命名为突变单元。再用samtools从原始的bam文件中提取突变位点上下游各250bp的所有reads,突变单元在这些reads中的位置应该是从左到右随机分布的,过滤掉分布异常的突变位点,比如突变单元全部集中在reads的左端或者右端。非致病位点过滤:对mutect2分析结果的位点进行注释(如annovar/snpeff/vep等注释软件)过滤掉位于基因间区、内含子非剪切区域、utr区域的突变位点;过滤提前准备好的人群频率数据库中的突变位点;过滤突变丰度低于百分之一且不在靶点数据库中的突变位点。噪音过滤:过滤噪音数据库中的突变位点,过滤位于str数据库中且突变丰度较低的突变位点。其中,str区域位点的过滤,还要结合肿瘤类型,msi状态,肿瘤样本中其它致病突变的丰度等多个因素进行综合考量,如某个msi-h的结直肠癌患者,有几个高丰度(如30%左右)的msh2基因移码突变,还有其它基因str区域丰度较低(5%左右)的移码突变,这些低丰度的移码突变就有很大可能是真的,具体还要再看umi的质控和突变单元的分布情况;
10.步骤四:靶点回捞:靶点数据库中的突变位点,如果不在此次检测结果中,则用samtools工具从原始的bam文件中提取对应坐标上下游250bp的所有reads,验证是否有该突变,以防低频的真实突变被分析软件因为样本降解、丰度太低、背景噪音过多而过滤掉。
11.作为本发明的一种优选实施方式,所述步骤一中的靶点数据库准备,收集nccn、fda、nmpa等机构推荐或获批的靶向药物相关突变位点;clinvar、oncokb、cosmic等数据库中致病或可能致病的突变位点,需要提取原始的bam文件再次进行验证,以防低频的真实突变被分析软件过滤掉。
12.作为本发明的一种优选实施方式,所述步骤一中的人群频率数据库准备,收集整理dbsnp、千人基因组、gnomad、exac等数据库中,人群频率大于千分之一的突变位点。
13.作为本发明的一种优选实施方式,所述步骤一中的噪音数据库准备,整理本地肿瘤数据库中高频出现的非致病突变位点,按照突变丰度的波动范围进行分类,挑出胚系突变、试剂和系统噪音突变,把这部分突变归类为噪音突变,后续直接过滤掉这部分突变位点。
14.作为本发明的一种优选实施方式,所述步骤一中的str数据库准备,收集人类基因组中str(短串联重复)区域重复单元的插入或缺失突变,这部分突变在肿瘤样本中大部分
是滑链错配导致的低频噪音,少部分是真实突变(如结直肠癌的msi-h患者),这部分突变不能直接按照噪音过滤掉,而要根据肿瘤类型、突变丰度等多个指标进行综合判断。
15.作为本发明的一种优选实施方式,所述步骤二中的复杂突变位点合并,对于复杂突变位点(如egfr 19del的delins突变),mutect2等分析软件并不是直接给出复杂突变的结果,而是两个或多个独立的突变,为了和靶点数据库相匹配,需要将其进行合并。
16.作为本发明的一种优选实施方式,所述步骤三中包括umi过滤、链偏好性过滤、突变单元分布过滤、非致病位点过滤和噪音过滤;
17.所述umi过滤从原始的bam文件中提取每个突变位点上下游各250bp范围的所有reads,对各个reads的umi进行统计,过滤测序错误引起的低频噪音突变,以及某个测序片段偏好性扩增引起的噪音突变;
18.所述链偏好性过滤,ngs杂交捕获测序过程中,来自正链的reads数目和来自负链的reads数目应该是对称的,分别统计来自正链的的未突变reads数、来自负链的未突变reads数、来自正链的突变reads数目、来自负链的突变reads数目,用费希尔精确检验计算其p值,过滤p值小于0.05的极端链偏好性突变。这部分噪音突变主要由于样本的氧化或者脱氨损伤、pcr扩增偏好等因素引起;
19.所述突变单元分布过滤,以突变位点为中心,左右各延伸10bp,把这段长约20bp的片段命名为突变单元。再从原始的bam文件中提取突变位点上下游各250bp的所有reads,突变单元在这些reads中的位置应该是从左到右随机分布的,过滤掉分布异常的突变位点(如偏好性低集中在所有reads的左端或者右端)。这部分噪音突变主要由于样本降解、测序仪在reads的端部测序质量较差、比对错误等原因引起;
20.所述非致病位点过滤,过滤基因间区、内含子非剪切区域、utr区域的突变;过滤人群频率大于千分之一的突变;过滤突变丰度低于百分之一且不在靶点数据库中的突变;
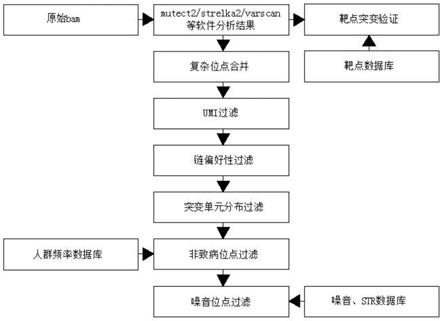
21.所述噪音位点过滤,过滤噪音数据库中的突变位点,过滤位于str数据库中且突变丰度较低的突变位点(还要结合肿瘤类型,msi状态等指标综合考量)。
22.作为本发明的一种优选实施方式,所述步骤四中的靶点回捞,靶点数据库中,不在检测结果里的突变位点,从原始的bam文件中提取相关reads,以防低频的真实突变被分析软件因为样本降解、丰度太低、背景噪音过多而过滤掉。
23.与现有技术相比,本发明的有益效果如下:
24.1.本发明能够显著降低肿瘤基因检测结果的噪音,呈现真实有效的突变位点,提高肿瘤基因检测的准确性,并降低生信分析人员的时间成本。
附图说明
25.通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
26.图1为本发明ngs肿瘤基因突变检测降噪方法的流程图。
具体实施方式
27.请参阅图1,本发明提供一种技术方案:ngs肿瘤基因突变检测降噪方法,包括以下步骤:
28.步骤一:提前准备好四个数据库:靶点数据库、人群频率数据库、噪音数据库、str数据库。靶点数据库的收集主要包括两个方面,一个是从nccn、fda、nmpa等机构发布的文档或者其官方文档中整理,另一个是下载clinvar、oncokb、cosmic数据库中致病或可能致病的突变位点(不包含短串联重复区域的突变),最后编写脚本(python/r等编程语言)将这两部分的突变位点合并,并以hgvs规则进行注释,以ghgvs作为每个突变位点的标签。人群频率数据库的准备,需要下载dbsnp、千人基因组、gnomad、exac等数据库的vcf文件,编写脚本提取人群频率大于千分之一(只要有一个数据库满足条件即可)的突变位点,同样以ghgvs作为每个突变位点的标签。噪音数据库依赖本地已分析的肿瘤样本数据,样本数量越多越好。其中高频出现(大于1%)的非致病突变位点,按照突变丰度的波动范围(突变丰度的最小值到最大值)进行分类,一般胚系突变的波动范围在40%~60%或者90%~100%,试剂和系统噪音突变一般在0~5%或者在某个恒定的值(如某个噪音突变在收录的肿瘤样本中都以20%左右的突变丰度出现)。把这两部分突变整理合并,并同样以ghgvs作为每个突变位点的标签。str数据库,编程提取人类基因组或某个bed区间内str(短串联重复)区域重复单元的插入或缺失突变,同样以ghgvs作为每个突变位点的标签;
29.步骤二:复杂位点合并:这里以一个复杂的egfr 19del突变举例,mutect2等分析软件分析出chr7:g.55242469_55242477del和chr7:g.55242478g》c这两个突变,这两个突变的丰度都在32%左右,而且通过查看bam文件,这两个突变发生在相同的reads上,所有这两个突变其实是一个复杂突变,需要将其合并,最终合并成chr7:g.55242469_55242478delinsc;
30.步骤三:假阳性过滤:这一步主要包含umi过滤、链偏好性过滤、突变单元分布过滤、非致病位点过滤、噪音过滤五个小步骤。umi过滤:用samtools工具从原始的bam文件中提取每个突变位点上下游各250bp范围的所有reads,对各个reads的umi进行统计,最终统计出每个umi所拥有的reads数(筛选大于等于2个reads支持的umi,短串联重复区域突变的阈值提高到3),及其在突变reads和未突变reads中的比例(过滤同时出现在未突变reads中的umi),筛选后的umi数大于等于3的突变位点可以保留。链偏好性过滤:mutect2等软件一般也有这方面的过滤,只是mutect2的过滤过于严苛,很多时候会过滤掉真实突变。本发明的方法是把mutect2分析结果中的sb值提取出来,再用r语言中的fisher.test函数计算链偏好性p值,过滤p值小于0.05的突变位点。突变单元分布过滤:以突变位点为中心,左右各延伸10bp,把这段长约20bp的片段命名为突变单元。再用samtools从原始的bam文件中提取突变位点上下游各250bp的所有reads,突变单元在这些reads中的位置应该是从左到右随机分布的,过滤掉分布异常的突变位点,比如突变单元全部集中在reads的左端或者右端。非致病位点过滤:对mutect2分析结果的位点进行注释(如annovar/snpeff/vep等注释软件)过滤掉位于基因间区、内含子非剪切区域、utr区域的突变位点;过滤提前准备好的人群频率数据库中的突变位点;过滤突变丰度低于百分之一且不在靶点数据库中的突变位点。噪音过滤:过滤噪音数据库中的突变位点,过滤位于str数据库中且突变丰度较低的突变位点。其中,str区域位点的过滤,还要结合肿瘤类型,msi状态,肿瘤样本中其它致病突变的丰度等多个因素进行综合考量,如某个msi-h的结直肠癌患者,有几个高丰度(如30%左右)的msh2基因移码突变,还有其它基因str区域丰度较低(5%左右)的移码突变,这些低丰度的移码突变就有很大可能是真的,具体还要再看umi的质控和突变单元的分布情况;
31.步骤四:靶点回捞:靶点数据库中的突变位点,如果不在此次检测结果中,则用samtools工具从原始的bam文件中提取对应坐标上下游250bp的所有reads,验证是否有该突变,以防低频的真实突变被分析软件因为样本降解、丰度太低、背景噪音过多而过滤掉。
32.本实施例中请参阅图1,所述步骤一中的靶点数据库准备,收集nccn、fda、nmpa等机构推荐或获批的靶向药物相关突变位点;clinvar、oncokb、cosmic等数据库中致病或可能致病的突变位点,需要提取原始的bam文件再次进行验证,以防低频的真实突变被分析软件过滤掉。
33.本实施例中请参阅图1,所述步骤一中的人群频率数据库准备,收集整理dbsnp、千人基因组、gnomad、exac等数据库中,人群频率大于千分之一的突变位点。
34.本实施例中请参阅图1,所述步骤一中的噪音数据库准备,整理本地肿瘤数据库中高频出现的非致病突变位点,按照突变丰度的波动范围进行分类,挑出胚系突变、试剂和系统噪音突变,把这部分突变归类为噪音突变,后续直接过滤掉这部分突变位点。
35.本实施例中请参阅图1,所述步骤一中的str数据库准备,收集人类基因组中str(短串联重复)区域重复单元的插入或缺失突变,这部分突变在肿瘤样本中大部分是滑链错配导致的低频噪音,少部分是真实突变(如结直肠癌的msi-h患者),这部分突变不能直接按照噪音过滤掉,而要根据肿瘤类型、突变丰度等多个指标进行综合判断。
36.本实施例中请参阅图1,所述步骤二中的复杂突变位点合并,对于复杂突变位点(如egfr 19del的delins突变),mutect2等分析软件并不是直接给出复杂突变的结果,而是两个或多个独立的突变,为了和靶点数据库相匹配,需要将其进行合并。
37.本实施例中请参阅图1,所述步骤三中包括umi过滤、链偏好性过滤、突变单元分布过滤、非致病位点过滤和噪音过滤;
38.所述umi过滤从原始的bam文件中提取每个突变位点上下游各250bp范围的所有reads,对各个reads的umi进行统计,过滤测序错误引起的低频噪音突变,以及某个测序片段偏好性扩增引起的噪音突变;
39.所述链偏好性过滤,ngs杂交捕获测序过程中,来自正链的reads数目和来自负链的reads数目应该是对称的,分别统计来自正链的的未突变reads数、来自负链的未突变reads数、来自正链的突变reads数目、来自负链的突变reads数目,用费希尔精确检验计算其p值,过滤p值小于0.05的极端链偏好性突变。这部分噪音突变主要由于样本的氧化或者脱氨损伤、pcr扩增偏好等因素引起;
40.所述突变单元分布过滤,以突变位点为中心,左右各延伸10bp,把这段长约20bp的片段命名为突变单元。再从原始的bam文件中提取突变位点上下游各250bp的所有reads,突变单元在这些reads中的位置应该是从左到右随机分布的,过滤掉分布异常的突变位点(如偏好性低集中在所有reads的左端或者右端)。这部分噪音突变主要由于样本降解、测序仪在reads的端部测序质量较差、比对错误等原因引起;
41.所述非致病位点过滤,过滤基因间区、内含子非剪切区域、utr区域的突变;过滤人群频率大于千分之一的突变;过滤突变丰度低于百分之一且不在靶点数据库中的突变;
42.所述噪音位点过滤,过滤噪音数据库中的突变位点,过滤位于str数据库中且突变丰度较低的突变位点(还要结合肿瘤类型,msi状态等指标综合考量)。
43.本实施例中请参阅图1,所述步骤四中的靶点回捞,靶点数据库中,不在检测结果
里的突变位点,从原始的bam文件中提取相关reads,以防低频的真实突变被分析软件因为样本降解、丰度太低、背景噪音过多而过滤掉。
44.在ngs肿瘤基因突变检测降噪方法使用的时候,提前准备好四个数据库:靶点数据库、人群频率数据库、噪音数据库、str数据库。靶点数据库的收集主要包括两个方面,一个是从nccn、fda、nmpa等机构发布的文档或者其官方文档中整理,另一个是下载clinvar、oncokb、cosmic数据库中致病或可能致病的突变位点(不包含短串联重复区域的突变),最后编写脚本(python/r等编程语言)将这两部分的突变位点合并,并以hgvs规则进行注释,以ghgvs作为每个突变位点的标签。人群频率数据库的准备,需要下载dbsnp、千人基因组、gnomad、exac等数据库的vcf文件,编写脚本提取人群频率大于千分之一(只要有一个数据库满足条件即可)的突变位点,同样以ghgvs作为每个突变位点的标签。噪音数据库依赖本地已分析的肿瘤样本数据,样本数量越多越好。其中高频出现(大于1%)的非致病突变位点,按照突变丰度的波动范围(突变丰度的最小值到最大值)进行分类,一般胚系突变的波动范围在40%~60%或者90%~100%,试剂和系统噪音突变一般在0~5%或者在某个恒定的值(如某个噪音突变在收录的肿瘤样本中都以20%左右的突变丰度出现)。把这两部分突变整理合并,并同样以ghgvs作为每个突变位点的标签。str数据库,编程提取人类基因组或某个bed区间内str(短串联重复)区域重复单元的插入或缺失突变,同样以ghgvs作为每个突变位点的标签,复杂位点合并:这里以一个复杂的egfr 19del突变举例,mutect2等分析软件分析出chr7:g.55242469_55242477del和chr7:g.55242478g》c这两个突变,这两个突变的丰度都在32%左右,而且通过查看bam文件,这两个突变发生在相同的reads上,所有这两个突变其实是一个复杂突变,需要将其合并,最终合并成chr7:g.55242469_55242478delinsc,这一步主要包含umi过滤、链偏好性过滤、突变单元分布过滤、非致病位点过滤、噪音过滤五个小步骤。umi过滤:用samtools工具从原始的bam文件中提取每个突变位点上下游各250bp范围的所有reads,对各个reads的umi进行统计,最终统计出每个umi所拥有的reads数(筛选大于等于2个reads支持的umi,短串联重复区域突变的阈值提高到3),及其在突变reads和未突变reads中的比例(过滤同时出现在未突变reads中的umi),筛选后的umi数大于等于3的突变位点可以保留。链偏好性过滤:mutect2等软件一般也有这方面的过滤,只是mutect2的过滤过于严苛,很多时候会过滤掉真实突变。本发明的方法是把mutect2分析结果中的sb值提取出来,再用r语言中的fisher.test函数计算链偏好性p值,过滤p值小于0.05的突变位点。突变单元分布过滤:以突变位点为中心,左右各延伸10bp,把这段长约20bp的片段命名为突变单元。再用samtools从原始的bam文件中提取突变位点上下游各250bp的所有reads,突变单元在这些reads中的位置应该是从左到右随机分布的,过滤掉分布异常的突变位点,比如突变单元全部集中在reads的左端或者右端。非致病位点过滤:对mutect2分析结果的位点进行注释(如annovar/snpeff/vep等注释软件)过滤掉位于基因间区、内含子非剪切区域、utr区域的突变位点;过滤提前准备好的人群频率数据库中的突变位点;过滤突变丰度低于百分之一且不在靶点数据库中的突变位点。噪音过滤:过滤噪音数据库中的突变位点,过滤位于str数据库中且突变丰度较低的突变位点。其中,str区域位点的过滤,还要结合肿瘤类型,msi状态,肿瘤样本中其它致病突变的丰度等多个因素进行综合考量,如某个msi-h的结直肠癌患者,有几个高丰度(如30%左右)的msh2基因移码突变,还有其它基因str区域丰度较低(5%左右)的移码突变,这些低丰度的移码突变就有
很大可能是真的,具体还要再看umi的质控和突变单元的分布情况;靶点回捞:靶点数据库中的突变位点,如果不在此次检测结果中,则用samtools工具从原始的bam文件中提取对应坐标上下游250bp的所有reads,验证是否有该突变,以防低频的真实突变被分析软件因为样本降解、丰度太低、背景噪音过多而过滤掉。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。