1.本发明涉及一种提高阳离子单体转化率的方法及聚合物微球乳液的制备方法。
背景技术:
2.聚合物微球作为一种深部调剖堵水剂越来越受到人们认可,其优点包括受外界影响小,可以直接用污水配制并在线注入,耐高温高盐,以及注入低粘度、无污染、成本较低等。该技术的设计机理是依靠纳米/微米级遇水可膨胀微球来逐级封堵地层孔喉实现其深部调剖堵水的效果。
3.油田所用的微球调驱剂一般指含交联剂的聚丙烯酰胺类球状微粒,按照粒径的大小,一般用反相微乳液聚合法制备纳米级微球、反相乳液法制备亚微米级微球、反相悬浮法制备微米至毫米级的微球。经过多年研究,人们通过配方、工艺等改进优化使聚合物微球从低固含量、均相结构向高固含量、非均相核壳结构方向发展,使得微球的耐温抗盐性能和调剖封堵效果大为提高,其中核壳型微球的研究成为研究热点。但经文献检索发现,大多研究者虽然采取了阴阳离子单体分步加料的聚合工艺,但对每一步反应中间产物的性质如单体转化率、电荷分布等表征分析不够,最终制得的微球不能确定其是阴阳核壳型,有可能是阴阳两性均相或互穿型、过渡型微球,这会造成微球封堵机理的改变、影响应用效果。
4.所谓的阴阳核壳型聚合物微球,一般指聚合物的核带明显的正电荷,采用丙烯酰胺类非离子单体与阳离子单体共聚而成,壳是丙烯酰胺类非离子单体与阴离子单体共聚而成。由于微球外壳带负电,因此可以克服地层的吸附作用、顺利进入油藏深部,在高温作用下,微球一边膨胀一边外壳逐步水解降解,暴露出带正电的微球内核,正负电荷的作用使得微球聚集成体积较大的团聚体,大大提高了封堵效果。也有研究者发现均相型阳离子纳米微球,在注入高渗油藏的过程中,一方面微球表面与带负电的孔隙壁存在静电吸引作用、增加吸附和滞留,一方面可以与聚驱后残留的聚合物产生吸附,提高流体的黏度并增加滞留。无论是哪种调剖机理,制备阳离子型聚合物微球都具有非常高的应用价值。
技术实现要素:
5.本发明所要解决的技术问题之一是现有技术中存在的采用反相微乳液、反相乳液或反相微悬浮聚合制备离子型聚合物微球过程中阳离子共聚单体转化率低、残留单体高的问题;如果继续进行阴离子包壳反应,会造成后续阴离子单体被电荷中和、阴离子度降低,使得阴阳核壳微球变成了类似于两性均相或互穿型、过渡型聚合物微球,改变了其封堵机理、降低封堵效果的问题。本发明通过在反相微乳液、反相乳液或反相微悬浮法制备阳离子聚合物微球过程中添加了少量转化助剂同时采用偶氮引发剂加氧化还原引发剂复合引发体系,较好的解决了上述技术问题,使得阳离子单体的转化率至接近100%。
6.本发明所要解决的技术问题之二是提供一种与解决技术问题之一相对应的阳离子聚合物微球乳液的制备方法。
7.本发明所要解决的技术问题之三是提供一种与解决技术问题之一相对应的阳离
子聚合物微球乳液。
8.为了解决以上技术问题,本发明的第一方面提供了一种提高阳离子单体转化率的方法,所述方法包括使非离子单体、阳离子单体在复合引发剂和转化助剂的存在下进行反应,其中所述复合引发剂包括氧化还原引发剂和偶氮引发剂,所述转化助剂选自二元羧酸类化合物中的一种或多种,所述转化助剂与所述非离子单体的摩尔比为1:(40-99),例如1:42、1:45、1:47、1:52、1:55、1:57、1:60、1:63、1:65、1:67、1:70、1:73、1:75、1:77、1:80、1:82、1:85、1:88、1:91、1:95、1:97以及它们之间的任意值。
9.根据本发明的一些实施方式,所述转化助剂与所述非离子单体的摩尔比为1:(50-90)。
10.根据本发明的一些实施方式,所述二元羧酸类化合物选自式(i)所示化合物中的一种或多种,
[0011][0012]
r1选自c1-c8的亚烃基,优选选自c1-c6的亚烷基或c2-c6的亚烯基,更优选选自亚甲基、亚乙基、亚丙基、亚丁基、亚乙烯基或亚丙烯基,
[0013]
r2选自c1-c6的烷基,优选选自c1-c4的烷基,更优选选自甲基、乙基或丙基,
[0014]
n为0或1。
[0015]
根据本发明的一些实施方式,所述二元羧酸类化合物选自乙二酸、丙二酸、丁二酸、己二酸、顺丁烯二酸、衣康酸和富马酸中的一种或多种。
[0016]
根据本发明的一些实施方式,所述转化助剂与阳离子单体的摩尔比为1:(1-20),例如1:2、1:4、1:5、1:6、1:7、1:8、1:9、1:11、1:13、1:15、1:17以及它们之间的任意值。
[0017]
根据本发明的一些实施方式,所述转化助剂与阳离子单体的摩尔比为1:(3-10)。
[0018]
现有技术中,分阶段补加活性单体am,可以提高dmdaac的聚合活性、从而使其参与共聚的效率提高;水溶性偶氮引发剂与无机过氧化物和其它水引发剂相比较,能进行平滑、稳定、可控制的分解反应,产生高线性和高分子量的聚合物,其分解特点是几乎全部为一级反应,只形成一种自由基,无诱导分解,并且对溶剂和杂质不敏感,由于引发速率较快,不具备氧化-还原性,可降低聚合过程中因离子化反应引起的体系ph值变化,提高体系中阳离子单体的竞聚率;也有在聚合过程中引入低分子量聚丙烯酸作为模板剂来提高阳离子单体竞聚率的方法,还有研究表明,微乳液可使单体间的活性差值缩小,从而使dmdaac在聚合物中的含量得到提高,阳离子度增大。但几乎没有提及若在单体中引入少量二元羧酸类化合物,与dmdaac电荷中心的阳离子部分中和后,可以降低电荷排斥作用,增加其双键的反应活性,从而提高共聚转化率。
[0019]
本发明在阳离子共聚反应中引入少量二元羧酸类化合物,一是与阳离子电荷中心的阳离子部分中和后,可以降低电荷排斥作用,增加阳离子单体双键的反应活性,从而提高共聚转化率,二是可以提高总电荷密度、增加静电力;同时采用偶氮引发剂加氧化还原引发剂复合引发体系,分段引发聚合、进一步提高阳离子单体的转化率至接近100%。
[0020]
根据本发明的一些实施方式,所述非离子单体选自带酰胺键的非离子单体中的一
种或多种。
[0021]
根据本发明的一些实施方式,所述非离子单体选自丙烯酰胺、甲基丙烯酰胺、n-异丙基丙烯酰胺、n,n-二甲基丙烯酰胺、n,n-二乙基丙烯酰胺、n-羟甲基丙烯酰胺、n-乙烯基甲酰胺和n-乙烯基乙酰胺中一种或多种。
[0022]
根据本发明的一些实施方式,所述阳离子单体选自季铵型阳离子单体中的一种或多种。
[0023]
根据本发明的一些实施方式,所述阳离子单体选自二甲基二烯丙基氯化铵、丙烯酰氧乙基三甲基氯化铵、甲基丙烯酰氧乙基三甲基氯化铵和甲基丙烯酰胺丙基三甲基氯化铵中一种或多种。
[0024]
根据本发明的一些实施方式,所述偶氮类引发剂选自偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁脒盐酸盐和偶氮二异丁咪唑啉盐酸盐中的一种或多种。
[0025]
根据本发明的一些实施方式,所述氧化还原引发剂中的氧化剂选自过硫酸钾、过硫酸钠、过硫酸铵和过氧化苯甲酰中的一种或多种;所述氧化还原引发剂中的还原剂选自亚硫酸钠、亚硫酸钾,亚硫酸氢钠、亚硫酸氢钾、硫代硫酸钠和氯化亚铁中的一种或多种。
[0026]
为了解决以上技术问题,本发明的第二方面提供了一种阳离子聚合物微球乳液的制备方法,所述方法包括使油相与水相在复合引发剂存在的条件下进行反应,
[0027]
所述油相包括油性液体与乳化剂,所述水相包括非离子单体、阳离子单体、转化助剂、助剂和水,所述复合引发剂包括氧化还原引发剂和偶氮引发剂,所述转化助剂选自二元羧酸类化合物中的一种或多种,所述转化助剂与所述非离子单体的摩尔比为1:(40-99),例如1:42、1:45、1:47、1:52、1:55、1:57、1:60、1:63、1:65、1:67、1:70、1:73、1:75、1:77、1:80、1:82、1:85、1:88、1:91、1:95、1:97以及它们之间的任意值。
[0028]
根据本发明的一些实施方式,所述转化助剂与所述非离子单体的摩尔比为1:(50-90)。
[0029]
根据本发明的一些实施方式,所述制备方法包括以下步骤:
[0030]
s1,将油性液体与乳化剂混合,得到油相;
[0031]
s2,将非离子单体、阳离子单体、转化助剂、助剂和水混合,得到水相;
[0032]
s3,将s1的油相、s2的水相和氧化还原引发剂混合,反应得到所述阳离子聚合物微球乳液,
[0033]
其中,当偶氮引发剂选自油溶性偶氮引发剂时,将所述油溶性偶氮引发剂与油性液体和乳化剂混合,得到油相;
[0034]
当偶氮引发剂选自水溶性偶氮引发剂时,将所述水溶性偶氮引发剂与非离子单体、阳离子单体、转化助剂、助剂和水混合,得到水相。
[0035]
根据本发明的一些实施方式,所述二元羧酸类化合物选自式(i)所示化合物中的一种或多种,
[0036][0037]
r1选自c1-c8的亚烃基,优选选自c1-c6的亚烷基或c2-c6的亚烯基,更优选选自亚
甲基、亚乙基、亚丙基、亚丁基、亚乙烯基或亚丙烯基,
[0038]
r2选自c1-c6的烷基,优选选自c1-c4的烷基,更优选选自甲基、乙基或丙基,
[0039]
n为0或1;
[0040]
根据本发明的一些实施方式,所述二元羧酸类化合物选自乙二酸、丙二酸、丁二酸、己二酸、顺丁烯二酸、衣康酸和富马酸中的一种或多种。
[0041]
根据本发明的一些实施方式,所述非离子单体选自带酰胺键的非离子单体中的一种或多种。
[0042]
根据本发明的一些实施方式,所述非离子单体选自丙烯酰胺、甲基丙烯酰胺、n-异丙基丙烯酰胺、n,n-二甲基丙烯酰胺、n,n-二乙基丙烯酰胺、n-羟甲基丙烯酰胺、n-乙烯基甲酰胺和n-乙烯基乙酰胺中一种或多种。
[0043]
根据本发明的一些实施方式,所述阳离子单体选自季铵型阳离子单体中的一种或多种。
[0044]
根据本发明的一些实施方式,所述阳离子单体选自二甲基二烯丙基氯化铵、丙烯酰氧乙基三甲基氯化铵、甲基丙烯酰氧乙基三甲基氯化铵和甲基丙烯酰胺丙基三甲基氯化铵中一种或多种。
[0045]
根据本发明的一些实施方式,述所述偶氮类引发剂选自偶氮二异丁腈、偶氮二异庚腈、偶氮二异丁脒盐酸盐和偶氮二异丁咪唑啉盐酸盐中的一种或多种。
[0046]
根据本发明的一些实施方式,所述氧化还原引发剂中的氧化剂选自过硫酸钾、过硫酸钠、过硫酸铵和过氧化苯甲酰中的一种或多种;所述氧化还原引发剂中的还原剂选自亚硫酸钠、亚硫酸钾,亚硫酸氢钠、亚硫酸氢钾、硫代硫酸钠和氯化亚铁中的一种或多种。
[0047]
根据本发明的一些实施方式,所述油性液体选自白油、煤油、液体石蜡、溶剂油和环己烷中一种或多种。
[0048]
根据本发明的一些实施方式,所述乳化剂为本领域常规使用的乳化剂,优选所述乳化剂包括选自亲油的失水山梨醇单油酸酯乳化剂、亲水的聚氧乙烯失水山梨醇脂肪酸酯、脂肪醇聚氧乙烯醚或异构脂肪醇醚乳化剂中的一种或两种以上的组合。
[0049]
根据本发明的一些实施方式,所述乳化剂的亲水亲油平衡值为4-8。
[0050]
根据本发明的一些实施方式,所述助剂包括助乳化剂、络合剂和交联剂中的一种或多种。
[0051]
根据本发明的一些实施方式,所述助乳化剂选自醇类或的盐类的助乳化剂,例如醋酸盐。
[0052]
根据本发明的一些实施方式,所述络合剂选自乙二胺四乙酸二钠和/或二乙烯三胺五乙酸钠。
[0053]
根据本发明的一些实施方式,所述交联剂选自亚甲基双丙烯酰胺、二乙烯基苯、聚乙二醇二丙烯酸酯和季戊四醇三丙烯酸酯中的一种或多种。
[0054]
根据本发明的一些实施方式,所述转化助剂与阳离子单体的摩尔比为1:(1-20),例如1:2、1:4、1:5、1:6、1:7、1:8、1:9、1:11、1:13、1:15、1:17以及它们之间的任意值。
[0055]
根据本发明的一些实施方式,所述转化助剂与阳离子单体的摩尔比1:(3-10)。
[0056]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述偶氮引发剂的质量含量为0.001-1%,例如0.005%、0.01%、0.03%、0.05%、
0.07%、0.1%、0.2%、0.3%、0.4%、0.7%、0.9%以及它们之间的任意值。
[0057]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述偶氮引发剂的质量含量为0.001-0.5%。
[0058]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述氧化还原引发剂的质量含量为0.001-1%,例如0.005%、0.01%、0.03%、0.05%、0.07%、0.1%、0.2%、0.3%、0.4%、0.7%、0.9%以及它们之间的任意值。
[0059]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述偶氮引发剂的质量含量为0.001-0.5%。
[0060]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述助乳化剂的质量含量为0.5-5%,例如1%、2%、3%、4%以及它们之间的任意值。
[0061]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述络合剂的质量含量为0.01-1%,例如0.1%、0.2%、0.3%、0.4%、0.7%、0.9%以及它们之间的任意值。
[0062]
根据本发明的一些实施方式,以所述非离子单体、阳离子单体和转化助剂的总重计,所述交联剂的质量含量为0.01-1%,例如0.1%、0.2%、0.3%、0.4%、0.7%、0.9%以及它们之间的任意值。
[0063]
根据本发明的一些实施方式,所述阳离子聚合物微球乳液的制备方法包括以下具体步骤:
[0064]
a)将油性液体和乳化剂体系混合均匀得到油相;
[0065]
b)将非离子单体、阳离子单体和转化助剂、助剂和水混合均匀得到水相,优选水相ph值为3~7;
[0066]
c)偶氮引发剂根据其是亲油还是亲水分别加入油相或水相中混匀,将水相与油相混合均匀,必要时均质乳化形成预乳液,再用氧化-还原引发剂引发反相微乳液、反相乳液或反相微悬浮聚合。
[0067]
根据本发明的一些实施方式,以重量份数计,所述油性液体的量为10~50份,乳化剂体系为1~20份,非离子单体、阳离子单体和转化助剂的总量为15~35份,助剂为0.5~5份,偶氮引发剂占非离子单体、阳离子单体和转化助剂的总量的0.001~1份,氧化还原引发剂占非离子单体、阳离子单体和转化助剂的总量的0.001~0.1份,其余部分为水。
[0068]
根据本发明的一些实施方式,所述阳离子聚合物微球乳液的制备方法包括以下具体步骤:
[0069]
a)将油性液体和乳化剂体系混合均匀得到油相,如采用油溶性偶氮引发剂则一并加入;
[0070]
b)将非离子单体、阳离子单体和转化助剂、助剂和水混合均匀得到水相,如采用水溶性偶氮引发剂则一并加入,优选水相ph值为3~7;
[0071]
c)将氧化-还原引发剂分别配制成水溶液待用;
[0072]
d)将水相与油相混合均匀,将乳液体系通惰性气体例如氮气除氧30~60min,然后加入氧化剂水溶液继续搅拌5-30min,优选10min,保持温度在5~35℃;加入优选滴加还原剂水溶液,当聚合温度上升到40~60℃时停止加入。继续保温反应1~3小时结束反应。
[0073]
根据本发明的一些实施方式,d)中如进行反相乳液聚合时需形成预乳液,优选用
均质机在10000~20000rpm下高速乳化2~10min形成预乳液。
[0074]
本发明中,采用反相(微)乳液、反相(微)悬浮的聚合方法制备阳离子聚合物微球过程中在阳离子共聚单体中引入少量二元羧酸类化合物,一是与阳离子电荷中心的阳离子部分中和后,可以降低电荷排斥作用,增加阳离子单体双键的反应活性,从而提高共聚转化率,二是可以提高总电荷密度、增加静电力;采用偶氮引发剂加氧化还原引发剂复合引发体系,分段引发聚合、进一步提高阳离子单体的转化率至接近100%,降低单体残留,无论是单独使用还是后续再进行包壳反应,都能使微球的性能特点得以完美体现。
[0075]
为了解决以上技术问题,本发明的第三方面提供了第二方面所述的方法制备的阳离子聚合物微球乳液。
[0076]
本发明的第四方面提供了一种核壳型聚合物微球,其包括第三方面所述的阳离子聚合物微球乳液与阴离子单体在引发剂存在下进行包壳反应的产物。
[0077]
本发明中所述“包壳反应”为本领域常规的操作,即将阴离子单体包覆在阳离子聚合物微球乳液周围,得到所述核壳型聚合物微球。
[0078]
根据本发明的一些实施方式,所述核壳型聚合物微球的制备方法包括:
[0079]
a)将油性液体和乳化剂体系混合均匀得到油相,如采用油溶性偶氮引发剂则一并加入;
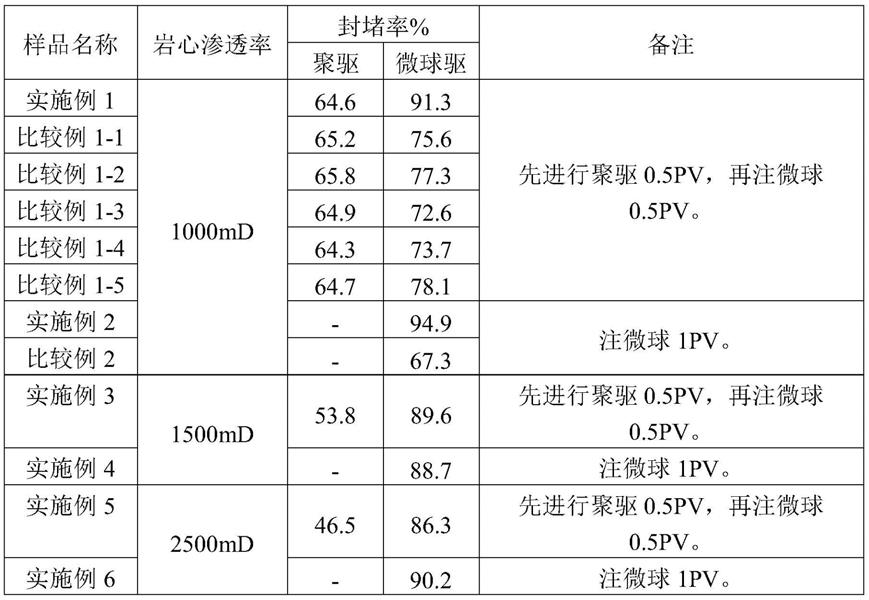
[0080]
b)将非离子单体、阳离子单体和转化助剂、助剂和水混合均匀得到水相,如采用水溶性偶氮引发剂则一并加入,优选水相ph值为3~7;
[0081]
c)将氧化-还原引发剂分别配制成水溶液待用;
[0082]
d)将水相与油相混合均匀,将乳液体系通惰性气体例如氮除氧30~60min,然后加入氧化剂水溶液继续搅拌5-30min,优选10min,保持温度在5~35℃;加入优选滴加还原剂水溶液,当聚合温度上升到40~60℃时停止加入。继续保温反应1~3小时结束反应,得阳离子聚合物微球乳液;
[0083]
e)将d)所得的阳离子聚合物微球乳液与包括阴离子单体和偶氮引发剂的水溶液混合进行包壳反应,优选在惰性气氛下如氮气下进行包壳反应,得到所述核壳型聚合物微球。
[0084]
根据本发明的一些实施方式,d)中如进行反相乳液聚合时需形成预乳液,优选用均质机在10000~20000rpm下高速乳化2~10min形成预乳液。
[0085]
根据本发明的一些实施方式,所述包括阴离子单体和偶氮引发剂的水溶液还包括非离子单体、助乳化剂、络合剂和交联剂中的一种或多种。
[0086]
根据本发明的一些实施方式,所述阴离子单体为本领域常规使用的阴离子单体,例如为含羧酸根或磺酸根的单体,如丙烯酸、甲基丙烯酸、2-丙烯酰胺基-2-甲基丙磺酸、乙烯基苯磺酸和乙烯基磺酸中的至少一种。
[0087]
本发明的第五方面提供了第二方面所述的方法制备的阳离子聚合物微球乳液、第三方面所述的阳离子聚合物微球乳液或第四方面的核壳型聚合物微球在油田开采中的应用,尤其是作为聚合物调驱剂的应用。
[0088]
根据本发明的一些实施方式,所述应用根据应用需要,所述聚合物微球调驱剂体系可以单独使用、也可加入驱油用表面活性剂搅拌均匀,或与表面活性剂以段塞式分别注入。
[0089]
根据本发明的一些实施方式,所述聚合物微球可用于油田水驱或化学驱后深度调剖、堵水、驱油等三次采油提高采收率的现场应用。
具体实施方式
[0090]
下面通过实施例对本发明作进一步的阐述,但是需要指出的是,本发明的保护范围并不受这此限制,而是由权利要求书来确定。
[0091]
需要特别说明的是,在本说明书的上下文中公开的两个或多个方面(或实施方式)可以彼此任意组合,由此而形成的技术方案属于本说明书原始公开内容的一部分,同时也落入本发明的保护范围之内。
[0092]
【实施例1】
[0093]
在反应釜中先加入1200g白油、300g span80、60g tween80和0.15g偶氮二异庚腈搅拌至完全混合均匀作为油相,搅拌转速为500rpm。在另外的容器中加入560g水、600g丙烯酰胺、250g二甲基二烯丙基氯化铵(60%含量)、15g己二酸、30g醋酸钠,0.75g乙二胺四乙酸二钠和3.75g亚甲基双丙烯酰胺搅拌溶解均匀作为水相,其中所述转化助剂己二酸和非离子单体丙烯酰胺的摩尔比为1:84.5,所述转化助剂己二酸和阳离子单体二甲基二烯丙基氯化铵的摩尔比为1:9.09;另外配制5%的氧化剂过硫酸铵和5%的还原剂亚硫酸氢钠各20ml。将全部水相加入反应釜内的油相中,一边搅拌一边通氮气40min,温度为22℃。滴入7.5g氧化剂水溶液,搅拌10min后再滴入还原剂8.0g,温度在几分钟内上升至82℃,保温反应1.5小时,然后降至30℃结束反应。
[0094]
取样分析固含量为25.1%,与理论值25.3%相比得出单体总转化率为99.2%;将微球乳液提纯后的干粉分析,zeta电位测得为35.8mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为98.8%。
[0095]
采用malvern zetasizer nano zs纳米粒度分析仪,用环己烷将微球乳液稀释成0.1%的溶液,搅拌5min并超声分散5min后在25℃下测试,取三次结果平均值作为微球的初始粒径。阳离子微球的粒径均值为118nm。
[0096]
【比较例1-1】
[0097]
水相中不加己二酸,其余与实施例1相同。
[0098]
取样分析固含量,与理论值相比得出单体总转化率为84.3%,zeta电位测得为12.7mv,xps测得阳离子转化率为45.6%。粒径仪测得粒径为112nm。
[0099]
【比较例1-2】
[0100]
油相中不加偶氮二异庚腈,其余与实施例1相同。
[0101]
取样分析固含量,与理论值相比得出单体总转化率为86.7%,zeta电位测得为13.3mv,xps测得阳离子转化率为52.5%。粒径仪测得粒径为115nm。
[0102]
【比较例1-3】
[0103]
水相中不加己二酸、油相中不加偶氮二异庚腈,其余与实施例1相同。
[0104]
取样分析固含量,与理论值相比得出单体总转化率为71.6%,zeta电位测得为9.2mv,xps测得阳离子转化率为29.3%。粒径仪测得粒径为104nm。
[0105]
【比较例1-4】
[0106]
水相中加入213g丙烯酰胺以及15g己二酸,即所述转化助剂己二酸和非离子单体
丙烯酰胺的摩尔比为1:30,其余与实施例1相同。取样分析固含量,与理论值相比得出单体总转化率为78.3%,zeta电位测得为8.2mv,xps测得阳离子转化率为45.2%。粒径仪测得粒径为107nm。
[0107]
【比较例1-5】
[0108]
水相中加入7.2g丙烯酸,即采用丙烯酸代替己二酸作为转化助剂,其中所述转化助剂丙烯酸和非离子单体丙烯酰胺的摩尔比为1:84.5,所述转化助剂丙烯酸和阳离子单体二甲基二烯丙基氯化铵的摩尔比为1:9.09,其余与实施例1相同。
[0109]
取样分析固含量,与理论值相比得出单体总转化率为87.6%,zeta电位测得为15.7mv,xps测得阳离子转化率为36.2%。粒径仪测得粒径为116nm。
[0110]
由以上结果可知,聚合体系中同时添加了己二酸和偶氮二异庚腈,对提高dmdaac的转化率效果显著。
[0111]
将六个微球乳液样品都以0.5%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0112]
【实施例2】
[0113]
在反应釜中先加入1200g环己烷、300gspan60、90gtween60和0.12g偶氮二异丁腈搅拌至完全混合均匀作为油相,搅拌转速为500rpm。在另外的容器中加入680g水、620g丙烯酰胺、200g二甲基二烯丙基氯化铵(80%含量)、15g乙二酸、30g醋酸钾、0.78g乙二胺四乙酸二钠和2.3g亚甲基双丙烯酰胺搅拌溶解均匀作为水相,其中所述转化助剂乙二酸和非离子单体丙烯酰胺的摩尔比为1:52.4,所述转化助剂乙二酸和阳离子单体二甲基二烯丙基氯化铵的摩尔比为1:5.94;另外配制5%的氧化剂过硫酸铵和5%的还原剂亚硫酸氢钠各20ml。将全部水相加入反应釜内的油相中,一边搅拌一边通氮气40min,温度为22℃。滴入7.2g氧化剂水溶液,搅拌10min后再滴入还原剂8.5g,温度很快上升至86℃,保温反应1.5小时,然后降至30℃结束第一步成核反应。
[0114]
取样分析固含量为25.1%,与理论值25.4%相比得出单体总转化率为98.8%,将微球乳液提纯后的干粉分析,zeta电位测得为32.3mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为98.3%。
[0115]
采用malvern zetasizer nano zs纳米粒度分析仪,用环己烷将微球乳液稀释成0.1%的溶液,搅拌5min并超声分散5min后在25℃下测试,取三次结果平均值作为微球的初始粒径。阳离子微球的粒径均值为126nm。
[0116]
另外配制包壳用水相溶液:在另外的容器中加入280g水、320g丙烯酰胺、50g苯乙烯磺酸钠、12g醋酸钾、0.28g乙二胺四乙酸二钠、1.3g亚甲基双丙烯酰胺、0.8g聚乙二醇二丙烯酸酯和0.08g v50搅拌溶解均匀。将该水溶液加入聚合物核乳液中,一边搅拌一边通氮气30min,温度为26℃。滴入10.8g氧化剂水溶液,搅拌10min后再滴入还原剂溶液12.5g,温度很快上升至76℃,保温反应1.5小时,然后降至30℃结束第二步包壳反应。
[0117]
取样分析固含量为30.6%,与理论值30.7%相比得出单体总转化率为99.7%,将微球乳液提纯后的干粉分析,zeta电位测得为-18.9mv,xps几乎测不出氯离子含量。说明微球外壳几乎不含阳离子,而基本为阴离子包覆。
[0118]
采用malvern zetasizer nano zs纳米粒度分析仪,用环己烷将微球乳液稀释成
0.1%的溶液,搅拌5min并超声分散5min后在25℃下测试,取三次结果平均值作为微球的初始粒径。微球的粒径均值为138nm。
[0119]
将微球乳液样品以0.5%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0120]
【比较例2】
[0121]
在实施例2的第一步成核反应中不添加乙二酸,其余步骤完全相同。
[0122]
取样分析固含量为21.3%,与理论值25.4%相比得出单体总转化率为83.9%,将微球乳液提纯后的干粉分析,zeta电位测得为23.6mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为52.3%。
[0123]
采用malvern zetasizer nano zs纳米粒度分析仪,用环己烷将微球乳液稀释成0.1%的溶液,搅拌5min并超声分散5min后在25℃下测试,取三次结果平均值作为微球的初始粒径。阳离子微球的粒径均值为117nm。
[0124]
第二步的包壳反应与实施例2完全相同。
[0125]
取样分析固含量为30.4%,与理论值30.7%相比得出单体总转化率为99.0%,将微球乳液提纯后的干粉分析,zeta电位测得为7.6mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为98.3%。
[0126]
采用malvern zetasizer nano zs纳米粒度分析仪,用环己烷将微球乳液稀释成0.1%的溶液,搅拌5min并超声分散5min后在25℃下测试,取三次结果平均值作为微球的初始粒径。微球的粒径均值为135nm。
[0127]
将微球乳液样品以0.5%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0128]
【实施例3】
[0129]
在3l玻璃烧杯中先加入700g白油、70g span80和10g aeo9,搅拌至完全混合均匀作为油相。在另外的容器中加入600g水、650g丙烯酰胺、200g二甲基二烯丙基氯化铵(60%含量)、20g顺丁烯二酸、20g醋酸钾、,0.8g乙二胺四乙酸二钠、2.5g亚甲基双丙烯酰胺和0.12g v50搅拌溶解均匀作为水相,其中所述转化助剂顺丁烯二酸和非离子单体丙烯酰胺的摩尔比为1:52.4,所述转化助剂顺丁烯二酸和阳离子单体二甲基二烯丙基氯化铵的摩尔比为1:4.27;另外配制2%的氧化剂过硫酸铵和0.05%的还原剂亚硫酸氢钠各20ml。将全部水相加入大烧杯内的油相中,用高速剪切乳化机在10000rpm下乳化10min,然后将乳液倒入聚合釜中,一边搅拌一边通氮气30min,搅拌转速为500rpm,温度为19℃。滴入3.2g氧化剂水溶液,搅拌10min后再滴入还原剂,控制滴加速度使升温速度保持在0.1~1℃/min,约3h后温度上升至56℃,停止滴加保温反应1.5小时,然后降至30℃结束反应。
[0130]
取样分析固含量为34.8%,与理论值35.0%相比得出单体总转化率为99.4%,将微球乳液提纯后的干粉分析,zeta电位测得为32.6mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为99.2%。
[0131]
采用malvern zetasizer nano zs纳米粒度分析仪,用白油将微球乳液稀释成0.1%的溶液,搅拌30min并超声分散5min后在40℃下测试,取三次结果平均值作为微球的
初始粒径。阳离子微球的粒径均值为356nm。
[0132]
将微球乳液样品以0.3%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0133]
【实施例4】
[0134]
在3l玻璃烧杯中先加入780g白油、70g span80和6g aeo20,搅拌至完全混合均匀作为油相。在另外的容器中加入500g水、550g丙烯酰胺、150g二甲基二烯丙基氯化铵(60%含量)、20g衣康酸、10g醋酸钾、0.6g乙二胺四乙酸二钠、1.5g季戊四醇三丙烯酸酯和0.12g v044搅拌溶解均匀作为水相,其中所述转化助剂衣康酸和非离子单体丙烯酰胺的摩尔比为1:51.6,所述转化助剂衣康酸和阳离子单体二甲基二烯丙基氯化铵的摩尔比为1:3.73;另外配制2%的氧化剂过硫酸铵和0.05%的还原剂亚硫酸氢钠各20ml。将全部水相加入大烧杯内的油相中,用高速剪切乳化机在15000rpm下乳化5min,然后将乳液倒入聚合釜中,一边搅拌一边通氮气30min,搅拌转速为500rpm,温度为21℃。滴入2.2g氧化剂水溶液,搅拌10min后再滴入还原剂,控制滴加速度使升温速度保持在0.1~1℃/min,约2h后温度上升至46℃,停止滴加保温反应1.5小时待用。
[0135]
取样分析固含量为31.3%,与理论值31.6%相比得出单体总转化率为99.1%,将微球乳液提纯后的干粉分析,zeta电位测得为28.3mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为98.9%。
[0136]
采用malvern zetasizer nano zs纳米粒度分析仪,用白油将微球乳液稀释成0.1%的溶液,搅拌30min并超声分散5min后在40℃下测试,取三次结果平均值作为微球的初始粒径。阳离子微球的粒径均值为292nm。
[0137]
另外配制包壳用水相溶液:在另外的容器中加入220g水、180g丙烯酰胺、60g 2-丙烯酰胺基-2-甲基丙磺酸钠;9g醋酸钠,0.16g乙二胺四乙酸二钠,0.7g亚甲基双丙烯酰胺,0.3g聚乙二醇二丙烯酸酯,0.06g v044搅拌溶解均匀。将该水溶液以滴加方式加入聚合物核乳液中,约1.5h加完,保温反应1小时,然后降至30℃结束第二步包壳反应。
[0138]
取样分析固含量为35.1%,与理论值35.2%相比得出单体总转化率为99.7%,将微球乳液提纯后的干粉分析,zeta电位测得为-22.3mv,xps几乎测不出氯离子含量。
[0139]
采用malvern zetasizer nano zs纳米粒度分析仪,用白油将微球乳液稀释成0.1%的溶液,搅拌30min并超声分散5min后在40℃下测试,取三次结果平均值作为微球的初始粒径。微球的粒径均值为382nm。
[0140]
将微球乳液样品以0.3%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0141]
【实施例5】
[0142]
在5l聚合釜中加入1200g溶剂油、102g span85和8g aeo15,搅拌至完全混合均匀作为连续相作为油相;水相ⅰ的组成如下:950g水、920g丙烯酰胺、300g丙烯酰氧乙基三甲基氯化铵、20g富马酸、乙二胺四乙酸二钠0.9g、醋酸钠30g、亚甲基双丙烯酰胺3.1g和0.08gv50搅拌溶解均匀,其中所述转化助剂富马酸和非离子单体丙烯酰胺的摩尔比为1:76.2,所述转化助剂富马酸和阳离子单体二甲基二烯丙基氯化铵的摩尔比为1:9.12;另外,
分别配制3%氧化剂过硫酸铵、1%还原剂亚硫酸氢钠备用。将水相ⅰ加入存放油相的聚合釜中搅拌15分钟至均匀,控制釜内温度为22℃,搅拌转速为500rpm。通氮气40min后先加入4.2毫升氧化剂溶液,搅拌15min后再滴入还原剂溶液8.6毫升,约15min后温度到达最高峰52℃,保温继续反应,约10分钟后聚合温度持续升高至79℃,保温反应1h后得到乳白色的聚合物微球乳液。
[0143]
取样分析固含量为31.3%,与理论值31.7%相比得出单体总转化率为98.7%,将微球乳液提纯后的干粉分析,zeta电位测得为29.6mv,xps测得氯离子含量再与投料中的理论值相比得到阳离子转化率为98.3%。
[0144]
采用malvern zetasizer nano zs纳米粒度分析仪,用白油将微球乳液稀释成0.1%的溶液,搅拌30min并超声分散5min后在40℃下测试,取三次结果平均值作为微球的初始粒径。阳离子微球的粒径均值为1562nm。
[0145]
将微球乳液样品以0.3%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0146]
【实施例6】
[0147]
在实施例5的基础上进行包壳反应。配制水相ⅱ、组成如下:450g水、520g丙烯酰胺、30g 2-丙烯酰氨基-2-甲基丙磺酸钠、20g苯乙烯、乙二胺四乙酸二钠0.4g、醋酸钠12g、亚甲基双丙烯酰胺0.5g、二乙烯基苯1.1g和0.18g v50,搅拌溶解均匀、调节ph=7.5作为水相ⅱ。将该水相加入实施例5反应好的微球乳液中,保持50℃,继续搅拌通氮,10min后温度开始上升,25min后达到最高温86℃,保温反应1h后得到半透明的聚合物微球乳液。
[0148]
取样分析固含量为36.8%,与理论值37.0%相比得出单体总转化率为99.5%,将微球乳液提纯后的干粉分析,zeta电位测得为-16.6mv,xps几乎测不出氯离子含量。
[0149]
采用malvern zetasizer nano zs纳米粒度分析仪,用白油将微球乳液稀释成0.1%的溶液,搅拌30min并超声分散5min后在40℃下测试,取三次结果平均值作为微球的初始粒径。微球的粒径均值为1865nm。
[0150]
将微球乳液样品以0.3%的浓度配制在总矿化度180000mg/l、钙 镁5700mg/l的模拟盐水中,放入95℃老化烘箱进行老化实验,3天后取出进行岩心注入实验,并计算封堵率,结果见表1。
[0151]
将上诉样品统一老化3天后取出,用φ25*300的人造岩心进行注入实验。先用相应的模拟盐水注入2pv,然后注入聚合物溶液和微球溶液各0.5pv,或只注微球溶液1pv,最后注入后续水驱至压力基本平稳。得出不同的注入压力,分别计算得到阻力系数和残余阻力系数,再由残余阻力系数换算出封堵率。
[0152]
表1.封堵实验结果汇总
[0153][0154]
聚合物选用0.1%疏水缔合聚合物(按cn104448128a实施例1制得)。
[0155]
由表1可见,实施例1、3、5采用的是阳离子型聚合物微球,粒径分别为纳米、亚微米和微米级,在聚驱后注入产生了很好的封堵作用;实施例2、4、6采用的是阳核阴壳型聚合物微球,直接注入也有很好的封堵作用。而比较例1-1、1-2、1-3虽然也是阳离子微球,但制备过程中没有添加阴离子单体或/和偶氮引发剂,造成阳离子单体转化率低,总的固含量低,这样的微球对同样的岩心封堵作用就下降较多;比较例2由于微球核的制备过程中阳离子单体转化率不高,造成包壳后虽然总的单体转化率较高,但是壳中的阴离子单体被核中残留的阳离子单体中和,造成最终的电性仍为正电荷,类似于一种阴阳互穿的两性微球,注入岩心后封堵率不高。从注入后的岩心端面可见,有一层凝胶糊在端面,没有进入到岩心内部,起不到深部调剖的作用。
[0156]
因此,本发明通过采用反相微乳液、反相乳液或反相微悬浮法制备阳离子聚合物微球过程中,水相中除了含丙烯酰胺类非离子单体及阳离子单体之外,还添加了少量阴离子单体,一是与阳离子电荷中心的阳离子部分中和后,可以降低电荷排斥作用,增加阳离子单体双键的反应活性,从而提高共聚转化率,二是可以提高总电荷密度、增加静电力;采用偶氮引发剂加氧化还原引发剂复合引发体系,分段引发聚合、进一步提高阳离子单体的转化率至接近100%,降低单体残留。无论是单独使用还是后续再进行包壳反应,都能使微球的性能特点得以完美体现。
[0157]
应当注意的是,以上所述的实施例仅用于解释本发明,并不对本发明构成任何限制。通过参照典型实施例对本发明进行了描述,但应当理解为其中所用的词语为描述性和解释性词汇,而不是限定性的词汇。可以按规定在本发明权利要求的范围内对本发明作出修改,以及在不背离本发明的范围和精神内对本发明进行修订。尽管其中描述的本发明涉及特定的方法、材料和实施例,但是并不意味着本发明限于其中公开的特定例,相反,本发
明可以扩展至其它所有具有相同功能的方法和应用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
