1.本发明属于药物化学技术领域,具体地说,涉及一种海绵环肽类化合物及其制备方法与应用。
背景技术:
2.环肽类化合物目前在药物治疗中取得了巨大成功,迄今为止,总共有超过40种环肽类药物正在临床应用中,平均每年约有一种环肽类药物进入治疗市场。值得注意的是,越来越多来自植物、动物和微生物的环肽被发掘并鉴定。比较有名的天然来源的环肽药物主要包括从真菌中分离的免疫抑制药物环孢菌素(cyclosporine);来源于海洋芋螺的镇痛药齐考诺肽(ziconotide);细菌来源的组蛋白脱乙酰基酶(hdac)抑制剂罗米地辛(romidepsin)以及从海鞘中分离得到抗肿瘤药物普利肽新(plitidepsin)。
3.海绵是海洋活性环肽的重要来源,自上世纪九十年代起,海绵来源的天然环肽呈现爆发式增长。已报道的海绵来源的环肽主要来自phakellia、axinella、hymeniacidon、callyspongia、stylissa等属的海绵,产生了phakellistatins、axinellins、hymenamides、stylissamides、axinastatin等类型环肽。环肽类化合物作为药物分子,具有抗癌、抗病毒、抗菌、免疫抑制等广泛的生物活性。因此,针对环肽类药物的研究正在受到人们越来越多的关注。
4.近年来,由于质谱技术的快速发展,极大推动了海洋天然产物的发现速度。特别是复合型液相串联三重四极杆质谱(q-q-q),通过母离子扫描、子离子扫描、中性丢失扫描和多离子反应监测等功能,大大提高了从复杂天然提取物中发现微量活性成分的灵敏度及选择性。基于环肽类化合物二级碎片离子的特点建立环肽类特征结构的质谱快速定位方法,快速筛选出复杂提取物中含有该骨架片段的新颖环肽类化合物,这将为海洋来源复杂化合物的相关研究提供新的研究思路,加快海洋药物开发的速度。
技术实现要素:
5.本发明的第一目的是提供一种海绵环肽类化合物。
6.本发明的第二目的是提供一种所述海绵环肽类化合物的制备方法。
7.本发明的第三目的是提供一种所述海绵环肽类化合物在制备免疫抑制药物中的应用。
8.为了实现上述目的,本发明采用的技术方案如下:
9.本发明的第一方面提供了一种海绵环肽类化合物,是一种具有免疫抑制活性的环肽,含有以下蛋氨酸亚砜片段:
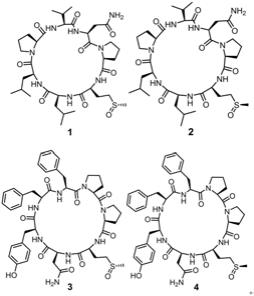
10.11.结构为以下化合物1~4中的一种:
[0012][0013]
本发明的第二方面提供了一种所述海绵环肽类化合物的提取方法,包括以下步骤:
[0014]
第一步,提取:将海绵粉碎后,用有机溶剂渗漉提取,合并提取液后减压浓缩得到提取液;
[0015]
第二步,萃取分离:提取液用等体积乙酸乙酯萃取3~5次(优选3次),合并乙酸乙酯层后浓缩得到的浸膏混悬于80~90%(优选85~90%)的甲醇水中,用等体积的石油醚萃取3~5次,将80~90%(优选85~90%)的甲醇水层稀释至50~60%的甲醇水,用等体积的二氯甲烷萃取3~5次(优选3次),合并萃取液并减压浓缩得到二氯甲烷萃取部分;
[0016]
第三步,分离富集:将上述二氯甲烷萃取部分通过sephadex lh-20凝胶柱层析,用50%的ch2cl
2-meoh洗脱剂洗脱,并采用质谱定位追踪的方式,将大分子量的化合物进行富集;采用ods中压柱色谱分离,用10%-100%的meoh/h2o梯度洗脱,采用质谱追踪定位分析,获得含有大分子量的环肽类化合物的精细馏分;
[0017]
第四步,筛选目标化合物:采用母离子扫描质谱法筛选含蛋氨酸亚砜的目标化合物,首先检测上述精细馏分中含碎片离子m/z 148的母离子m/z 781.55和913.92[m h]
,对其进行子离子扫描验证,二级碎片离子中能发现m/z 148的子离子,进而获得其碎片离子信息和色谱保留行为;
[0018]
第五步,质谱导向分离:在获得其碎片离子信息和色谱保留行为基础上,采用质谱导向的半制备高液相色谱法对上述精细馏分进行分离,得到目标化合物;
[0019]
第六步,非对映体拆分:目标化合物在液相色谱中呈现单一色谱峰,核磁数据分析证明其为在手性亚砜处的混合物;
[0020]
分离获得的分子离子781.55经半制备型超临界流体色谱法,在viridis beh色谱柱上实现基线分离,放大到半制备规模进行拆分,获得海绵环肽类化合物1和2;分子离子913.92在sfcib-3色谱柱上实现基线分离,放大到半制备规模进行拆分,获得海绵环肽类化合物3和4。
[0021]
所述第一步中的有机溶剂选自乙醇、甲醇、丙醇中的至少一种。
[0022]
所述第五步中,质谱导向的半制备高液相色谱法的分离条件:50-60%甲醇-水,流速5.0ml/min,在正离子模式下检测分子离子781.55和913.92。
[0023]
所述第六步中,在viridis beh色谱柱上实现基线分离的条件:30%甲醇-二氧化碳。
[0024]
所述第六步中,分子离子913.92在sfcib-3色谱柱上实现基线分离的条件:27%甲醇-二氧化碳。
[0025]
本发明的第三方面提供了一种所述海绵环肽类化合物的制备方法,包括以下步骤:
[0026]
[0027][0028]
第一步,按照肽链上的氨基酸序列,将第一种氨基酸即fmoc保护的脯氨酸装载到2-ctc树脂中:将2-ctc树脂在无水dcm中溶胀,加入fmoc保护的脯氨酸和diea(n,n-二异丙基乙胺),反应完全后洗涤去除未反应的原料;
[0029]
第二步,fmoc脱保护:在室温下,使用含15~30%(优选20%)哌啶的dmf溶液进行fmoc脱保护,然后将树脂用dmf洗涤;
[0030]
第三步,按照肽链上的氨基酸序列,依次通过以下步骤偶联其余氨基酸:将fmoc保护的氨基酸、hatu和diea在dmf中,室温下与树脂轻轻涡旋搅拌,然后将树脂用dmf洗涤;其余氨基酸中的酪氨酸和天冬酰胺的侧链分别由t-bu基团和trt基团保护;
[0031]
第四步,fmoc脱保护:在室温下,使用含15~30%(优选20%)哌啶的dmf溶液进行fmoc脱保护,然后将树脂用dmf洗涤;
[0032]
第五步,裂解:过滤树脂,并用hfip裂解溶液在无水dcm中裂解,裂解完成后纯化,得到侧链保护的线性肽4a;
[0033]
第六步,大环化:将摩尔比为1:(2~4):(2~4):(2~4)的侧链保护的线性肽4a、edci、hoat、diea溶于dcm中,室温搅拌反应(1~24h),然后真空除去溶剂;
[0034]
第七步,侧链脱保护:向第六步获得的含保护基的环肽中加入裂解混合物进行脱保护,反应完全后纯化得到化合物4。
[0035]
所述第一步具体的方法步骤如下:将1个当量的2-ctc树脂溶于无水dcm中溶胀15~30分钟,加入1.5~2.5个当量fmoc保护的脯氨酸和3~5个当量diea的dcm溶液,在室温下
涡旋摇动0.8~1.5小时,向反应混合物中加入meoh,并将树脂旋转10~20分钟,过滤树脂并依次用dcm、dcm/meoh(v/v=1∶1)和meoh洗涤;
[0036]
所述第三步具体步骤如下:按照肽链上的氨基酸序列,依次通过以下步骤偶联其余氨基酸:将2~4个当量的fmoc保护的氨基酸、2~4个当量的hatu和5~7个当量的diea在dmf中,室温下与树脂轻轻涡旋搅拌0.8~1.2小时,然后将树脂用dmf洗涤;其余氨基酸中的酪氨酸和天冬酰胺的侧链分别由t-bu基团和trt基团保护;
[0037]
所述第五步具体步骤如下:裂解:过滤树脂,并用15~25%的hfip裂解溶液在无水dcm中处理0.8~1.5小时,然后重复此步骤处理0.3~1小时;过滤后,合并所得裂解溶液,真空浓缩,通过hplc纯化,得到侧链保护的线性肽4a;
[0038]
所述第七步中的裂解混合物是由以下体积百分比的组分制成:体积百分比为90~95%的tfa(三氟乙酸)、体积百分比为2.5~5%的tis(三异丙基硅烷)和体积百分比为2.5~5%的h2o。
[0039]
优选的,所述第七步中的裂解混合物是由以下体积百分比的组分制成:体积百分比为90~95%的tfa(三氟乙酸)、体积百分比为2.5~3.5%的tis(三异丙基硅烷)和体积百分比为2.5~3.5%的h2o。
[0040]
本发明的第四方面提供了一种所述海绵环肽类化合物在制备免疫抑制药物中的应用。
[0041]
所述免疫抑制的细胞选自cd4
t细胞、b细胞、bmdm巨噬细胞。
[0042]
所述海绵环肽类化合物对cd4
t细胞、b细胞和bmdm巨噬细胞这三株免疫细胞显示出较强的抑制活性,并且不同亚砜构型的海绵环肽类化合物呈现出显著的活性差异。其中含s-亚砜的化合物1活性明显优于含r-亚砜的化合物2,也优于拆分前的混合物。而含r-亚砜的化合物4活性明显优于含s-亚砜的化合物3,也优于拆分前的混合物。这也说明含单一构型亚砜的环肽和非对映体混合物之间存在显著的活性差异。因此本发明化合物可用于制备免疫抑制药物。
[0043]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0044]
本发明提供的海绵环肽类化合物的制备方法简单,免疫抑制活性显著。本发明为研究和开发新免疫抑制药提供了新的先导化合物,为微量环肽类活性成分快速识别和定向追踪提供了新方法,为含手性亚砜环肽的拆分提供了新策略,为环肽类化合物的合成提供了新思路,为开发利用我国海洋药用资源提供了科学依据。
[0045]
本发明从轴海绵属海绵中分离获得一种具有免疫抑制活性的新环肽类化合物,并且运用超临界流体色谱技术对含蛋氨酸亚砜的非对映体环肽进行拆分,获得了单一构型纯的蛋氨酸亚砜环肽。本发明运用固相合成结合液相环化策略实现了对本发明化合物的全合成。
[0046]
本发明提供了一种从复杂提取物中选择性识别含蛋氨酸亚砜的环肽类化合物的质谱快速定位方法。从肽化合物的质谱裂解机制中发现,环肽经碰撞诱导解离后形成的二级碎片离子往往是组成该环肽的基本单元——各氨基酸残基的离子。根据这一特点,分析二级ms发现,可以通过碎片离子为m/z 148的母离子扫描法快速寻找含蛋氨酸亚砜的环肽。运用母离子质谱法扫描从中国西沙群岛附近海域采集的轴海绵属海绵axinella sp.的粗馏分,从中发现母离子为m/z 781.55和913.92[m h]
的峰,在获得其碎片离子信息和色谱
保留行为基础上,结合质谱引导制备液相将其分离,并运用超临界流体色谱拆分得到一种新的非对应纯的环肽类化合物。
[0047]
本发明运用固相合成及液相环化策略实现了对本发明化合物的全合成,为其后续研究提供了量的保证。
附图说明
[0048]
图1是海绵环肽类化合物1-4的发现和拆分流程示意图。
[0049]
图2是海绵环肽类化合物1和2的ms/ms碎片检测示意图。
[0050]
图3是海绵环肽类化合物3和4的ms/ms碎片检测示意图。
[0051]
图4是海绵环肽类化合物1的x-ray ortep示意图。
[0052]
图5是海绵环肽类化合物2的x-ray ortep示意图。
[0053]
图6是海绵环肽类化合物3的x-ray ortep示意图。
[0054]
图7是海绵环肽类化合物4的1h nmr(dmso-d6,600mhz)谱对比示意图。
[0055]
图8是海绵环肽类化合物4的
13
c nmr(dmso-d6,150mhz)谱对比示意图。
[0056]
图9是海绵环肽类化合物1-4对三种免疫细胞抑制结果示意图。
具体实施方式
[0057]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0058]
实施例1
[0059]
从海绵中发现并提取海绵环肽类化合物1-4
[0060]
从环肽化合物的质谱裂解机制中发现,环肽经碰撞诱导解离后形成的二级碎片离子往往是组成该环肽的基本单元——各氨基酸残基的离子。根据这一特点,分析二级ms发现,可以通过碎片离子为m/z 148的母离子扫描法快速寻找含蛋氨酸亚砜的环肽。运用母离子质谱法扫描从中国西沙群岛附近海域采集的轴海绵属海绵axinella sp.的粗馏分,从中发现母离子为m/z 781.55和913.92[m h]
的峰,如图1所示,在获得其碎片离子信息和色谱保留行为基础上,结合质谱引导制备液相将其分离,并运用超临界流体色谱拆分得到一种新的非对应纯的环肽类化合物,即海绵环肽类化合物1-4。
[0061]
母离子扫描质谱法在本发明化合物筛选中的应用具体如下:
[0062]
1)共有碎片离子的确定:
[0063]
结合含蛋氨酸亚砜环肽的二级质谱及蛋氨酸亚砜残基的结构,碎片离子为m/z 148,将“母离子扫描”方式的子离子设为148。
[0064]
2)“母离子扫描”的优化:
[0065]
输入电压、碰撞能量等的优化,获得尽可能多的高响应度碎片离子信息,并确保能够找到母离子,一般母离子为碎片离子响应度的1/3为宜;设定筛选标准,设定子离子为148作为标准,保证具有该特点的离子通过。
[0066]
3)样品前处理条件的优化:
[0067]
通过sephadex lh-20凝胶柱层析,用50%的ch2cl
2-meoh洗脱剂洗脱,并采用质谱
定位追踪的方式,将大分子量的化合物主要富集;经历反相中压柱色谱及质谱导向的全制备高液相色谱,有效的去除杂质成分,富集环肽类成分。
[0068]
4)液相条件的优化:
[0069]
为使获得的特异成分实现基线分离,减少离子间干扰,便于多级离子结构分析和特异性成分的定向获取,确定最佳的线性梯度条件,尽量避免共流出现象的发生。
[0070]
5)质谱引导制备分离:
[0071]
在获得其碎片离子信息和色谱保留行为基础上,结合质谱引导制备液相将m/z 781.55和913.92[m h]
分离。
[0072]
6)非对映体拆分:
[0073]
目标化合物m/z 781.55和913.92[m h]
在液相色谱中呈现单一色谱峰,核磁数据分析证明其为在手性亚砜处的混合物。在超临界流体色谱仪上筛选多种不同的固定相和改性剂后,目标化合物m/z 781.55和913.92[m h]
分别在viridis beh柱和ib-3柱上实现了基线分离。放大到半制备规模进行拆分,得到非对映纯的海绵环肽类化合物1-4。
[0074]
从海绵中提取海绵环肽类化合物1-4的具体步骤如下:
[0075]
第一步,提取:轴海绵属海绵(干重92g)(从中国西沙群岛附近海域采集)粉碎后,用95%的乙醇5.4l渗漉提取三次,合并提取液后减压浓缩,回收乙醇,浓缩得到提取液。
[0076]
第二步,萃取分离:提取液用等体积乙酸乙酯萃取三次,合并乙酸乙酯层后浓缩得到乙酸乙酯层浸膏(14.5g)。将乙酸乙酯层浸膏混悬于90%的甲醇水中,用等体积石油醚萃取三次,合并萃取液并减压浓缩得到石油醚部分(3.5g)。将90%的甲醇水层稀释至60%的甲醇水,用等体积的二氯甲烷萃取三次,合并萃取液并减压浓缩得到二氯甲烷部分(2.0g)。
[0077]
第三步,分离富集:将上述二氯甲烷萃取部分2.0g,通过sephadex lh-20凝胶柱层析,用50%的ch2cl
2-meoh洗脱剂洗脱,并采用质谱定位追踪的方式,将大分子量的化合物进行富集得1.3g。采用ods中压柱色谱分离,用meoh/h2o梯度(10%-100%,450min)洗脱,采用质谱追踪定位分析,获得含有大分子量的环肽类化合物的精细馏分;
[0078]
第四步,筛选目标化合物:采用母离子扫描质谱法筛选含蛋氨酸亚砜的目标化合物,首先检测馏分中含碎片离子m/z 148的母离子m/z 781.55和913.92[m h]
,对其进行子离子扫描验证,二级碎片离子中能发现m/z 148的子离子,进而获得其碎片离子信息和色谱保留行为;
[0079]
第五步,质谱导向分离:在获得其碎片离子信息和色谱保留行为基础上,采用质谱导向的半制备高液相色谱法对上述精细馏分进行分离(50-60%甲醇-水,流速5.0ml/min,在正离子模式下检测分子离子781.55和913.92)。
[0080]
第六步,非对映体拆分:目标化合物在液相色谱中呈现单一色谱峰,核磁数据分析证明其为在手性亚砜处的混合物;
[0081]
分离获得的分子离子781.55经半制备型超临界流体色谱法,在viridis beh色谱柱上实现基线分离(30%甲醇-二氧化碳),放大到半制备规模进行拆分,获得海绵环肽类化合物1和2。而分子离子913.92在sfcib-3色谱柱上实现基线分离(27%甲醇-二氧化碳),获得海绵环肽类化合物3和4。
[0082]
图1是海绵环肽类化合物1-4的发现和拆分流程示意图。其中,a是粗馏分fr.2.g的总离子流色谱图;b是经过母离子扫描锁定的含m/z 148片段的目标环肽色谱图;c是二级质
谱验证图;d和e分别是目标环肽m/z 781和913的拆分色谱图。从图中可以看出,经过母离子扫描结合超临界流体拆分技术成功从海绵粗提物中定向获得光学纯的含蛋氨酸亚砜的环肽类化合物。
[0083]
化合物1~4的结构如下所示:
[0084][0085]
通过上述步骤制得的海绵环肽类化合物1-4的理化性质和核磁共振数据如下:
[0086]
化合物1:无色晶体;mp 283-285℃;[α]
25d-57.2(c 0.29,meoh);ir(atr)ν
max 3280,2956,2924,2856,1672,1642,1617,1522,1453,1412,1344,1285,1257,1178,1099,1022,872,793cm-1
;hresims m/z 781.4279[m h]
(calcd for c
36h61
n8o9s,781.4282).
[0087]
化合物2:无色晶体;mp 226-228℃;[α]
25d-39.1(c 0.31,meoh);ir(atr)ν
max 3280,2956,2924,2856,1672,1642,1617,1522,1453,1412,1344,1285,1257,1178,1099,1022,872,793cm-1
;hresims m/z 781.4279[m h]
(calcd for c
36h61
n8o9s,781.4282).
[0088]
化合物3:无色晶体;mp 204-206℃;[α]
25d-15.8(c 0.28,meoh);uv(meoh)λ
max
(logε)277(3.08)nm;ir(atr)ν
max 3312,2923,2854,1729,1652,1616,1512,1444,1372,1347,1321,1237,1200,1101,1023,802,700cm-1
;hresims m/z 913.3917[m h]
(calcd for c
46h57
n8o
10
s,913.3918).
[0089]
化合物4:白色无定型粉末;[α]
25d-31.9(c 0.27,meoh);uv(meoh)λ
max
(logε)277(3.08)nm;ir(atr)ν
max 3312,2923,2854,1729,1652,1616,1512,1444,1372,1347,1321,1237,1200,1101,1023,802,700cm-1
;hresims m/z 913.3917[m h]
(calcd for c
46h57
n8o
10
s,913.3918).
[0090]
海绵环肽类化合物1-4的核磁共振谱数据见表1、表2。
[0091]
表1:化合物1和2的核磁共振谱数据(dmso-d6)
[0092]
[0093]
[0094][0095]
化合物1和2有相同的平面结构,通过仔细分析2d nmr(tocsy、cosy和hmbc)谱,可以确定组成它们的7个氨基酸残基包括一个缬氨酸、一个天冬酰胺、一个蛋氨酸亚砜、两个亮氨酸和两个脯氨酸。
[0096]
7个氨基酸残基的连接顺序,通过分析hmbc和roesy相关信号及esi-ms/ms确定。hmbc相关信号leu
2-nh/leu
1-co、leu
1-nh/meto-co、meto-nh/pro
1-co、asn-nh/val-co和val-nh/pro
2-co,确定了结构片段pro
1-meto-leu
1-leu2和pro
2-val-asn的存在。结合roesy相关信号leu
2-hα/pro
2-hα和asn-hα/pro
1-hδ,确定该环肽的结构为cyclo-(pro
1-meto-leu
1-leu
2-pro
2-val-asn)。通过esi-qtof-ms/ms质谱分析发现,b系碎片离子峰:m/z 667、568、471、358和245,对应母离子依次丢失asn、val、pro、leu和leu这些中性分子的碎片离子峰;相应地,y系碎片离子峰:m/z 684、537、424、311和214,对应母离子依次丢失pro、meto、leu、leu和pro,这证实了该环肽的nmr结构解析结果。如图2所示,图2是海绵环肽类化合物1和2的ms/ms碎片检测示意图。
[0097]
表2:化合物3和4的核磁共振谱数据(dmso-d6)
[0098]
[0099]
[0100][0101]
化合物3和4有相同的平面结构,通过仔细分析2d nmr(tocsy、cosy和hmbc)谱,可以确定组成该化合物的7个氨基酸残基包括一个酪氨酸、一个天冬酰胺、一个蛋氨酸亚砜、
两个苯丙氨酸和两个脯氨酸。
[0102]
7个氨基酸残基的连接顺序,通过分析hmbc和roesy相关信号及esi-ms/ms确定。hmbc相关信号asn-nh/meto-co、met(o)-nh/pro
2-co、phe
2-nh/phe
1-co和phe
1-nh/tyr-co,确定了结构片段pro
2-meto-asn和tyr-phe
1-phe2。结合roesy相关信号pro
1-hα/pro
2-hα、phe
2-hα/pro
1-hδ和asn-hα/tyr-nh,确定该环肽的结构为cyclo-(pro
1-pro
2-meto-asn-tyr-phe
1-phe2)。
[0103]
通过esi-qtof-ms/ms质谱分析发现,b系碎片离子峰:m/z 766、619、456、342和195,对应母离子依次丢失phe、phe、tyr、asn和met(o)这些中性分子的碎片离子峰;相应地,y系碎片离子峰:m/z 816、719、572、458和295,对应母离子依次丢失pro、pro、met(o)、asn、tyr和phe,这证实了该环肽的nmr结构解析结果(如图3所示,图3是海绵环肽类化合物3和4的ms/ms碎片检测示意图。)。
[0104]
化合物1-3的绝对构型通过x-ray单晶衍射技术确定,化合物1的x-ray ortep图如图4所示,图4是海绵环肽类化合物1的x-ray ortep示意图,其所有的氨基酸构型均为l型,蛋氨酸亚砜中硫的构型为s型;化合物2的x-ray ortep图如图5所示,图5是海绵环肽类化合物2的x-ray ortep示意图,其所有的氨基酸构型均为l型,蛋氨酸亚砜中硫的构型为r型。化合物3的x-ray ortep图如图6所示,图6是海绵环肽类化合物3的x-ray ortep示意图,其所有的氨基酸构型均为l型,蛋氨酸亚砜中硫的构型为r型。
[0105]
化合物1-3的单晶数据见表3:
[0106]
表3:化合物1-3的单晶数据
[0107]
[0108][0109]
实施例2
[0110]
化合物4的合成路线
[0111][0112]
第一步,按照肽链上的氨基酸序列,将第一种氨基酸即fmoc保护的脯氨酸装载到2-ctc树脂中:
[0113]
将2-ctc树脂(100mg,上样量:1.0mmol/g)在装有2ml无水dcm的一次性容器(torivq)中溶胀20分钟。加入fmoc-pro-oh(2.0当量)和diea(4.0当量)的dcm溶液,并将反应容器在室温下在涡旋中摇动1小时。向反应混合物中加入200μl meoh,并将树脂旋转15分钟。过滤树脂并用无水dcm(3ml
×
5次,1分钟/次),1:1dcm/meoh(v/v)(3ml
×
5次,1分钟/次)和meoh(3ml
×
2次,1分钟/次)洗涤。
[0114]
第二步,fmoc脱保护:在室温下,使用3ml含20%哌啶的dmf溶液进行fmoc脱保护20分钟,然后将树脂用dmf洗涤(3ml
×
2次,1分钟/次)。
[0115]
第三步,肽偶联:将所需的每种fmoc保护的氨基酸(3个当量),hatu(3个当量)(多肽缩合试剂,系统命名为2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)和diea(6个当量)在dmf中,室温下与树脂轻轻涡旋搅拌1小时,然后将树脂用dmf洗涤(3ml
×
5次,1分钟/次)。
[0116]
第四步,fmoc脱保护:在室温下,使用3ml含20%哌啶的dmf溶液进行fmoc脱保护20分钟,然后将树脂用dmf洗涤(3ml
×
2次,1分钟/次)。
[0117]
第五步,裂解:过滤树脂,并用3ml 20%(v/v)hfip裂解溶液在无水dcm中处理1小时,然后重复此步骤处理30分钟。过滤后,合并所得裂解溶液,真空浓缩,通过hplc纯化(waters xbridge c18,5μm,10
×
250mm,ch3cn/h2o=23/77,流速为5.0ml/min,tr为23.5min),得到侧链保护的线性肽4a。
[0118]
第六步,大环化:将第五步获得的侧链保护的线性肽4a(1当量)在dcm中的溶液加入edci(3当量)(1-乙基-3(3-二甲基丙胺)碳二亚胺)、hoat(3当量)(1-羟基-7-氮杂苯并三氮唑)和diea(3当量),将该溶液在室温搅拌16h,然后真空除去溶剂。
[0119]
第七步,侧链脱保护:向第六步获得的含保护基的环肽32.3mg中加入1ml裂解混合物(tfa:tis:h2o=95/2.5/2.5,v/v/v),搅拌3h,通过lc-ms(4.6
×
150mm,3.5μm;ch3cn/h2o=10/90到90/10,流速为1.0ml/min,tr为6.5min)监测反应。将该溶液真空浓缩,然后通过hplc纯化(waters xbridge c18,5μm,10
×
250mm,ch3cn/h2o=25/75,流速为5.0ml/min,tr为19.3min),得到化合物4(收率43%)。
[0120]
通过比较nmr数据,证明合成的化合物4同提取的化合物4完全匹配,如图7和图8所示。图7是海绵环肽类化合物4的1h nmr(dmso-d6,600mhz)谱对比示意图。图8是海绵环肽类化合物4的
13
c nmr(dmso-d6,150mhz)谱对比示意图。
[0121]
实施例3
[0122]
本发明化合物1-4的体外活性实验
[0123]
骨髓源巨噬细胞(bmdm)制备:4周龄c57雄性小鼠,脱颈处死后浸泡于装有75%酒精的烧杯内5分钟。用眼科剪在脚踝处环形切开,剪断跟腱,分离出胫骨末端,进一步分开股骨和胫骨,并去除肌肉、筋膜,完成粗分离,浸泡于装有5%双抗的pbs的培养皿中,剪开干骺端,冲洗骨髓腔,冲入新的培养皿中。收集冲洗出来的培养基悬液,1000rpm 5min离心,丢弃上清液,换含20%fbs、1%双抗、10ng/ml mcsf的高糖dmem重悬,5%co237度培养。使用f4/80抗体流式检测纯度,纯度>95%的细胞用于活性抑制试验。
[0124]
cd4
t细胞和b细胞制备:从雌性balb/c小鼠(18-20g)获取新鲜脾细胞,并将细胞在含10%fbs的rpmi 1640培养基中培养。使用cd4
t细胞分离试剂盒,从脾细胞单细胞悬浮液中分离cd4
t细胞,使用抗cd4-apc抗体染色cd4
t细胞。而b细胞是使用b细胞分离试剂盒从小鼠脾细胞悬浮液中进行分离,然后分别用cd45r(b220)-pe和抗生物素-apc进行荧光染色。cd4
t细胞和b细胞的纯度通过流式细胞术分析确定。
[0125]
细胞活力测试:本发明化合物1-4、1/2混合物和3/4混合测试了对cd4
t细胞、b细胞和bmdm细胞的抑制活性。样品用dmso溶解,低温保存,dmso在最终体系中的浓度控制在不影响检测活性的范围之内,倍比稀释为1-100μg/ml的工作浓度。取对数生长期细胞,制成单
细胞悬液1
×
106个/ml,将该悬液加到96孔板中,每孔加入100μl。于5%co2,37℃培养箱中培养24h后,分别加入10μg/ml的受试药物,每个样品均设3个复孔,阴性对照为等体积培养基及相应的dmso浓度为溶媒对照,以消除dmso对细胞生长的影响。于5%co2,37℃培养箱中培养48h后,每孔加入10μl ckk8溶液,继续培养4h后,在450nm处测定各孔吸光值(od值)。每个样品在测试中均设置复孔(n=3),在结果中以标准偏差(sd)表示。多组间比较采用anova分析,组间两两比较采用games-howell检验。
[0126]
如图9所示,图9是海绵环肽类化合物1-4对三种免疫细胞抑制结果示意图。从图中可以看出,化合物1-4对cd4
t、b和bmdm这三株免疫细胞具有抑制作用,并且不同亚砜构型的环肽化合物呈现出显著的活性差异。其中含s-亚砜的化合物1活性明显优于含r-亚砜的化合物2,也优于拆分前的混合物。而含r-亚砜的化合物4活性明显优于含s-亚砜的化合物3,也优于拆分前的混合物。这也说明含单一构型亚砜的环肽和非对映体混合物之间存在显著的活性差异。本发明化合物是潜在免疫抑制药物,为研制新的免疫药物提供了新的先导化合物。
[0127]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。