1.本发明涉及一种杂环类化合物及其制备方法和应用。
背景技术:
2.专利cn113087700a公开了2-((s)-4-((r)-4-氯-2
’‑
((四氢-1h-吡咯嗪-7a(5h)-基)甲氧基)-2,3,5’,8
’‑
四氢-6’h-螺[茚-1,7
’‑
喹唑啉]-4
’‑
基)哌嗪-2-基)乙腈(参照实施例49合成的化合物45),也公开了该化合物能够有效治疗癌症,例如肺癌、胰腺癌或结直肠癌。
[0003]
然而,药物活性成分可以以不同晶型存在,不同的晶型可能具有性质上的差异。不同晶型所导致的性质改变也可以改进最终的制剂形式,例如,这种改变可以增加溶解度,进而提高生物利用度,或者改善活性成分的稳定性,或者更为出人意料地,在增加溶解度的同时具有良好的稳定性。对于上述的2-((s)-4-((r)-4-氯-2
’‑
((四氢-1h-吡咯嗪-7a(5h)-基)甲氧基)-2,3,5’,8
’‑
四氢-6’h-螺[茚-1,7
’‑
喹唑啉]-4
’‑
基)哌嗪-2-基)乙腈,有必要对其不同晶型进行进一步地开发和改进。
技术实现要素:
[0004]
本发明所要解决的技术问题是为了克服2-((s)-4-((r)-4-氯-2
’‑
((四氢-1h-吡咯嗪-7a(5h)-基)甲氧基)-2,3,5’,8
’‑
四氢-6’h-螺[茚-1,7
’‑
喹唑啉]-4
’‑
基)哌嗪-2-基)乙腈2-((s)-4-((r)-4-氯-2
’‑
((四氢-1h-吡咯嗪-7a(5h)-基)甲氧基)-2,3,5’,8
’‑
四氢-6’h-螺[茚-1,7
’‑
喹唑啉]-4
’‑
基)哌嗪-2-基)乙腈缺乏可药用晶型的缺陷,改善其理化性质,从而提供了一种如结构式i化合物所示的杂环类化合物及其制备方法和应用。
[0005]
本发明提供了一种如式i所示的杂环类化合物,其为晶型、或者为药学上可接受的盐或溶剂合物的结晶或无定型:。
[0006]
其中,所述的药学上可接受的盐可为其盐酸盐、其硫酸盐、其马来酸盐、其磷酸盐、其富马酸盐、其甲磺酸盐、其草酸盐或其氢溴酸盐。
[0007]
(1)本发明提供了有一种如式i所示的化合物的游离碱晶型a,其以2θ角表示的x射线粉末衍射图,在13.303
±
0.2
º
、20.882
±
0.2
º
、15.601
±
0.2
º
、24.193
±
0.2
º
、25.479
±
0.2
º
、14.553
±
0.2
º
、16.654
±
0.2
º
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其以2θ角
表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:20.12
±
0.2
º
、16.033
±
0.2
º
、19.517
±
0.2
º
、10.461
±
0.2
º
、26.394
±
0.2
º
、7.81
±
0.2
º
、29.218
±
0.2
º
、7.759
±
0.2
º
、14.182
±
0.2
º
、22.249
±
0.2
º
、26.897
±
0.2
º
、28.909
±
0.2
º
、30.097
±
0.2
º
、27.076
±
0.2
º
、22.788
±
0.2
º
、11.222
±
0.2
º
、28.124
±
0.2
º
、11.691
±
0.2
º
、31.64
±
0.2
º
、33.075
±
0.2
º
、32.082
±
0.2
º
、12.169
±
0.2
º
和38.959
±
0.2
º
。
[0008]
在本发明一些实施例方案中,所述的如式i所示的化合物的游离碱晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表1所示。
[0009]
在本发明有一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其xrpd图谱基本如图6所示。
[0010]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其热重分析曲线在室温至148.3℃的温度范围失重为0.65%。
[0011]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其热重分析曲线(tga)图谱基本如图7所示。
[0012]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其差示扫描量热(dsc)曲线在121.63
±
5℃处具有吸热峰。
[0013]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其差示扫描量热图谱基本如图8所示。
[0014]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型a,其动态水吸附(dvs)图谱基本如图9所示。其dvs曲线在80%rh下的吸湿增重为1.82
±
0.02%(例如1.82%)。
[0015]
(2)本发明提供了一种有如式i所示化合物的游离碱晶型b,其以2θ角表示的x射线粉末衍射图,在13.326
±
0.2
º
、20.63
±
0.2
º
、16.826
±
0.2
º
、20.239
±
0.2
º
、6.758
±
0.2
°
、14.317
±
0.2
º
和8.492
±
0.2
º
处有衍射峰。
[0016]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型b,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:24.428
±
0.2
º
、25.364
±
0.2
º
和28.766
±
0.2
º
。
[0017]
在本发明一些实施例方案中,所述的如式i所示的化合物的游离碱晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表2所示。
[0018]
在本发明有一些实施方案中,所述的如式i所示的化合物的游离碱晶型b,其xrpd图谱基本如图11所示。
[0019]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型b,其热重分析曲线在室温至139℃的温度范围失重为3.96%。
[0020]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型b,其热重分析曲线(tga)图谱基本如图12所示。
[0021]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型b,其差示扫描量热(dsc)曲线在111.30
±
5℃处具有吸热峰。
[0022]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型b,其差示扫描量热图谱基本如图13所示。
[0023]
(3)本发明提供了一种有如式i所示化合物的游离碱晶型c,其以2θ角表示的x射线
粉末衍射图,在3.486
±
0.2
º
、6.739
±
0.2
º
、19.773
±
0.2
º
、16.501
±
0.2
º
、13.246
±
0.2
°
、24.273
±
0.2
º
和15.643
±
0.2
º
处有衍射峰。
[0024]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型c,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:29.609
±
0.2
º
、26.512
±
0.2
º
、20.882
±
0.2
º
、21.393
±
0.2
º
、25.266
±
0.2
º
、28.476
±
0.2
º
、22.253
±
0.2
º
、21.942
±
0.2
º
、23.765
±
0.2
º
、29.057
±
0.2
º
、25.481
±
0.2
º
、14.517
±
0.2
º
、31.306
±
0.2
º
、38.92
±
0.2
º
、32.585
±
0.2
º
和10.775
±
0.2
º
。
[0025]
在本发明一些实施例方案中,所述的如式i所示的化合物的游离碱晶型c,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表3所示。
[0026]
在本发明有一些实施方案中,所述的如式i所示的化合物的游离碱晶型c,其xrpd图谱基本如图14所示。
[0027]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型c,其热重分析曲线在室温至133.1℃的温度范围失重为6.75%。
[0028]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型c,其热重分析曲线图谱基本如图15所示。
[0029]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型c,其差示扫描量热曲线在70.59
±
5℃和124.14
±
5℃处具有吸热峰。
[0030]
在本发明一些实施方案中,所述的如式i所示的化合物的游离碱晶型c,其差示扫描量热图谱基本如图16所示。
[0031]
(4)本发明提供了有一种如式i所示的化合物的盐酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在6.5
±
0.2
º
、13.41
±
0.2
º
、17.91
±
0.2
º
、20.78
±
0.2
º
、23.78
±
0.2
º
、20.28
±
0.2
º
、18.44
±
0.2
º
和25.95
±
0.2
º
处有衍射峰。
[0032]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:21.67
±
0.2
º
、26.98
±
0.2
º
、30
±
0.2
º
、25.55
±
0.2
º
、16.24
±
0.2
º
、11.8
±
0.2
º
、9.45
±
0.2
º
和14.43
±
0.2
º
。
[0033]
在本发明一些实施例方案中,所述的如式i所示的化合物的盐酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表7所示。
[0034]
在本发明有一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其xrpd图谱基本如图17所示。
[0035]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其热重分析曲线在室温至160℃温度范围内失重8.93%。
[0036]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其热重分析曲线(tga)图谱基本如图18所示。
[0037]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其差示扫描量热(dsc)曲线在158.4℃
±
5℃处具有吸热峰。
[0038]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其差示扫描量热图谱基本如图19所示。
[0039]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型a,其动态水吸附(dvs)图谱基本如图21所示。其dvs曲线在80%rh下的吸湿增重为5.02%
±
0.02%(例如
5.02%)。
[0040]
(5)本发明提供了一种有如式i所示的化合物的盐酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在13.38
±
0.2
º
、6.51
±
0.2
º
、18.08
±
0.2
º
、18.42
±
0.2
º
、20.95
±
0.2
°
、26.09
±
0.2
º
、23.98
±
0.2
º
和20.11
±
0.2
º
处有衍射峰。
[0041]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型b时,2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:21.64
±
0.2
º
、30.25
±
0.2
º
、14.32
±
0.2
º
和16.29
±
0.2
º
。
[0042]
在本发明一些实施例方案中,所述的如式i所示的化合物的盐酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表8所示。
[0043]
在本发明有一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型b,其xrpd图谱基本如图22所示。
[0044]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型b,其热重分析曲线在室温至160℃温度范围内失重12.68%。
[0045]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型b,其热重分析曲线图谱基本如图23所示。
[0046]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型b,其差示扫描量热曲线在153.2℃
±
5℃和169.8℃
±
5℃处具有吸热峰。
[0047]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型b,其差示扫描量热图谱基本如图24所示。
[0048]
(6)本发明提供了一种有如式i所示的化合物的盐酸盐晶型c,其以2θ角表示的x射线粉末衍射图,在13.3
±
0.2
º
、18.39
±
0.2
º
、6.65
±
0.2
º
、20.01
±
0.2
º
、21.67
±
0.2
º
、14.22
±
0.2
º
和17.35
±
0.2
º
处有衍射峰。
[0049]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型c时,2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:22.22
±
0.2
º
、16.73
±
0.2
º
和26.7
±
0.2
º
。
[0050]
在本发明一些实施例方案中,所述的如式i所示的化合物的盐酸盐晶型c,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表9所示。
[0051]
在本发明有一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型c,其xrpd图谱基本如图26所示。
[0052]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型c,其热重分析曲线在室温至150℃温度范围内失重7.82%。
[0053]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型c,其热重分析曲线图谱基本如图27所示。
[0054]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型c,其差示扫描量热曲线在105.6℃
±
5℃、131.5℃
±
5℃和140.9℃
±
5℃处具有吸热峰。
[0055]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐晶型c,其差示扫描量热图谱基本如图28所示。
[0056]
(7)本发明提供了一种有如式i所示的化合物的盐酸盐无定型,其以2θ角表示的x射线粉末衍射图,其2θ角表示的x射线粉末衍射图基本如图30所示。
[0057]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐无定型,其热重分析曲线在室温至131℃温度范围内失重3.4%。
[0058]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐无定型,其热重分析曲线图谱基本如图31所示。
[0059]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐无定型,其玻璃化转变温度为101.66℃。
[0060]
在本发明一些实施方案中,所述的如式i所示的化合物的盐酸盐无定型,其差示扫描量热图谱基本如图32所示。
[0061]
(8)本发明提供了一种有如式i所示的化合物的硫酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在13.22
±
0.2
º
、6.95
±
0.2
º
、22.82
±
0.2
º
、18.89
±
0.2
º
、10.66
±
0.2
°
、15.12
±
0.2
º
、20.99
±
0.2
º
和17.07
±
0.2
º
处有衍射峰。
[0062]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a时,2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:17.45
±
0.2
º
、19.49
±
0.2
º
、13.95
±
0.2
º
、15.56
±
0.2
º
、25.47
±
0.2
º
、26.57
±
0.2
º
、24.49
±
0.2
º
和28.22
±
0.2
º
。
[0063]
在本发明一些实施例方案中,所述的如式i所示的化合物的硫酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表11所示。
[0064]
在本发明有一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a,其xrpd图谱基本如图33所示。
[0065]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a,其热重分析曲线在室温至150℃温度范围内失重6.52%。
[0066]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a,其热重分析曲线图谱基本如图34所示。
[0067]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a,其差示扫描量热曲线在108.7℃
±
5℃和148.3℃
±
5℃处具有吸热峰。
[0068]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a时,其差示扫描量热图谱基本如图35所示。
[0069]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型a时,其动态水分吸附图谱基本如图37所示。其dvs曲线在80%rh下的吸湿增重为11.01%
±
0.02%。
[0070]
(9)本发明提供了一种有如式i所示的化合物的硫酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在13.23
±
0.2
º
、22.66
±
0.2
º
、16.96
±
0.2
º
、25.35
±
0.2
º
、6.78
±
0.2
°
和28.38
±
0.2
º
处有衍射峰。
[0071]
在本发明一些实施例方案中,所述的如式i所示的化合物的硫酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表12所示。
[0072]
在本发明有一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型b,其xrpd图谱基本如图39所示。
[0073]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型b,其热重分析曲线在室温至150℃温度范围内失重7.93%。
[0074]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型b,其热重分析曲线图谱基本如图40所示。
[0075]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型b,其差示扫描量热曲线在96.5℃
±
5℃和142.9℃
±
5℃处具有吸热峰。
[0076]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型b,其差示扫描量热图谱基本如图41所示。
[0077]
(10)本发明提供了有一种如式i所示的化合物的硫酸盐晶型c,其以2θ角表示的x射线粉末衍射图,在22.41
±
0.2
º
、20.15
±
0.2
º
、6.7
±
0.2
º
、6.93
±
0.2
º
、19.81
±
0.2
º
、16.78
±
0.2
º
、13.34
±
0.2
º
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:21
±
0.2
º
、21.27
±
0.2
°
、18.56
±
0.2
º
、13.06
±
0.2
º
、25.47
±
0.2
º
、21.69
±
0.2
º
、10.04
±
0.2
º
、17.41
±
0.2
º
、26.54
±
0.2
º
、10.45
±
0.2
º
、23.11
±
0.2
º
、24.54
±
0.2
º
、13.98
±
0.2
º
、19.23
±
0.2
º
、29.02
±
0.2
°
、30.07
±
0.2
º
和15.44
±
0.2
º
。
[0078]
在本发明一些实施例方案中,所述的如式i所示的化合物的硫酸盐晶型c,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表13所示。
[0079]
在本发明有一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其xrpd图谱基本如图43所示。
[0080]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其热重分析曲线在室温至150℃温度范围内失重5.9%。
[0081]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其热重分析曲线(tga)图谱基本如图44所示。
[0082]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其差示扫描量热(dsc)曲线在148.5
ºc±
5℃处具有吸热峰。
[0083]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其差示扫描量热图谱基本如图45所示。
[0084]
在本发明一些实施方案中,所述的如式i所示的化合物的硫酸盐晶型c,其动态水吸附(dvs)图谱基本如图47所示。其dvs曲线在80%rh下的吸湿增重为3.72%
±
0.02%(例如3.72%)。
[0085]
(11)本发明提供了有一种如式i所示的化合物的马来酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在11.8
±
0.2
º
、10.79
±
0.2
º
、17.34
±
0.2
º
、12.54
±
0.2
º
、16.59
±
0.2
º
、7.59
±
0.2
º
和24.61
±
0.2
º
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:25.54
±
0.2
º
、19.63
±
0.2
º
、25.94
±
0.2
º
、14.92
±
0.2
º
、22.91
±
0.2
º
、13.36
±
0.2
º
、9.78
±
0.2
º
、10.43
±
0.2
º
、22.12
±
0.2
º
、18.8
±
0.2
º
、27.86
±
0.2
º
和21.09
±
0.2
°
。
[0086]
在本发明一些实施例方案中,所述的如式i所示的化合物的马来酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表15所示。
[0087]
在本发明有一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其xrpd图谱基本如图49所示。
[0088]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其热重
分析曲线在室温至130℃温度范围内失重9.1%。
[0089]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其热重分析曲线图谱基本如图50所示。
[0090]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其差示扫描量热曲线在95.0℃
±
5℃处具有吸热峰。
[0091]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其差示扫描量热图谱基本如图51所示。
[0092]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型a,其动态水吸附(dvs)图谱基本如图53所示。其dvs曲线在80%rh下的吸湿增重为5.93%
±
0.02%。
[0093]
(12)本发明提供了有一种如式i所示的化合物的马来酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在15.31
±
0.2
º
、17.24
±
0.2
º
、10.8
±
0.2
º
、20.34
±
0.2
º
、13.54
±
0.2
º
、14.46
±
0.2
º
、11.8
±
0.2
º
和7.84
±
0.2
º
处有衍射峰。
[0094]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型b,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:12.36
±
0.2
º
、12.95
±
0.2
º
、16.53
±
0.2
º
、25.36
±
0.2
º
、24.55
±
0.2
º
、19.59
±
0.2
º
、22.74
±
0.2
º
、17.87
±
0.2
°
和9.31
±
0.2
º
。
[0095]
在本发明一些实施例方案中,所述的如式i所示的化合物的马来酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表16所示。
[0096]
在本发明有一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型b,其xrpd图谱基本如图54所示。
[0097]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型b,其热重分析曲线在室温至130℃温度范围内失重5.0%。
[0098]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型b,其热重分析曲线图谱基本如图55所示。
[0099]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型b,其差示扫描量热曲线在92.4℃
±
5℃和126.7℃
±
5℃处具有吸热峰。
[0100]
在本发明一些实施方案中,所述的如式i所示的化合物的马来酸盐晶型b,其差示扫描量热图谱基本如图56所示。
[0101]
(13)本发明提供了有一种如式i所示的化合物的磷酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在13.03
±
0.2
º
、24.1
±
0.2
º
、21.13
±
0.2
º
、16.79
±
0.2
º
、16.23
±
0.2
°
、24.98
±
0.2
º
、19.55
±
0.2
º
和13.6
±
0.2
º
处有衍射峰。
[0102]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型a,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:26.12
±
0.2
º
、20.31
±
0.2
º
、15.23
±
0.2
º
、21.94
±
0.2
º
、22.82
±
0.2
º
、11.16
±
0.2
º
、26.6
±
0.2
º
和17.85
±
0.2
º
。
[0103]
在本发明一些实施例方案中,所述的如式i所示的化合物的磷酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表18所示。
[0104]
在本发明有一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型a,其xrpd图谱基本如图58所示。
[0105]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型a,其热重分
析曲线在室温至180℃温度范围内失重6.0%。
[0106]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型a,其热重分析曲线图谱基本如图59所示。
[0107]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型a,其差示扫描量热曲线在113.5℃
±
5℃和161.7℃
±
5℃处具有吸热峰。
[0108]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型a,其差示扫描量热图谱基本如图60所示。
[0109]
(14)本发明提供了有一种如式i所示的化合物的磷酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在5.74
±
0.2
º
、16.29
±
0.2
º
、17.64
±
0.2
º
、23.14
±
0.2
º
、20.73
±
0.2
°
、19.15
±
0.2
º
和12.68
±
0.2
º
处有衍射峰。
[0110]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型b,其2θ角表示的x射线粉末衍射图,还在7.65
±
0.2
º
和/或11.65
±
0.2
º
处有衍射峰。
[0111]
在本发明一些实施例方案中,所述的如式i所示的化合物的磷酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表19所示。
[0112]
在本发明有一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型b,其xrpd图谱基本如图62所示。
[0113]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型b,其热重分析曲线在室温至180℃温度范围内失重6.0%。
[0114]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型b,其热重分析曲线图谱基本如图63所示。
[0115]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型b,其差示扫描量热曲线在77.2℃
±
5℃和184.0℃
±
5℃处具有吸热峰。
[0116]
在本发明一些实施方案中,所述的如式i所示的化合物的磷酸盐晶型b,其差示扫描量热图谱基本如图64所示。
[0117]
(15)本发明提供了有一种如式i所示的化合物的富马酸盐无定型,其以2θ角表示的x射线粉末衍射图,其2θ角表示的x射线粉末衍射图基本如图66所示。
[0118]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐无定型时,其热重分析曲线在室温至163℃温度范围内失重6.0%。
[0119]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐无定型时,其热重分析曲线图谱基本如图67所示。
[0120]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐无定型时,其差示扫描量热曲线在53.5℃
±
5℃和101.4℃
±
5℃处具有吸热峰。
[0121]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐无定型时,其差示扫描量热图谱基本如图68所示。
[0122]
(16)本发明提供了有一种如式i所示的化合物的富马酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在24.43
±
0.2
º
、13.42
±
0.2
º
、16.65
±
0.2
º
、17.75
±
0.2
º
、13.56
±
0.2
º
、25.36
±
0.2
º
、18.49
±
0.2
º
和11.28
±
0.2
°
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:20.42
±
0.2
º
、22.89
±
0.2
º
、13.93
±
0.2
º
、21.11
±
0.2
º
、21.58
±
0.2
°
、17.35
±
0.2
º
、10.9
±
0.2
º
、27.93
±
0.2
°
、26.9
±
0.2
º
、15.03
±
0.2
º
和5.64
±
0.2
º
。
[0123]
在本发明一些实施例方案中,所述的如式i所示的化合物的富马酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,衍射峰和峰高百分比还可如表20所示。
[0124]
在本发明有一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其xrpd图谱基本如图70所示。
[0125]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其热重分析曲线在室温至150℃温度范围内失重2.65%。
[0126]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其热重分析曲线(tga)图谱基本如图71所示。
[0127]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其差示扫描量热(dsc)曲线在153.1
±
5℃处具有吸热峰。
[0128]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其差示扫描量热图谱基本如图72所示。
[0129]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型a,其动态水吸附(dvs)图谱基本如图74所示。其dvs曲线在80%rh下的吸湿增重为1.41%
±
0.02%(例如1.41%)。
[0130]
(17)本发明提供了有一种如式i所示的化合物的富马酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在14.103
±
0.2
º
、19.927
±
0.2
º
、26.22
±
0.2
º
、15.33
±
0.2
º
、6.68
±
0.2
º
、20.631
±
0.2
º
和16.342
±
0.2
º
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:24.371
±
0.2
º
、24.89
±
0.2
º
、21.136
±
0.2
º
、23.986
±
0.2
º
、22.459
±
0.2
º
、25.134
±
0.2
º
、27.776
±
0.2
º
、18.524
±
0.2
º
、6.941
±
0.2
º
、28.248
±
0.2
º
、30.875
±
0.2
º
、29.357
±
0.2
º
、6.276
±
0.2
º
、38.373
±
0.2
º
、13.651
±
0.2
º
、17.798
±
0.2
º
、31.401
±
0.2
º
、13.012
±
0.2
º
、4.966
±
0.2
º
和10.068
±
0.2
º
。
[0131]
在本发明一些实施例方案中,所述的如式i所示的化合物的富马酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表21所示。在本发明有一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其xrpd图谱基本如图76所示。
[0132]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其热重分析曲线在室温至142.9
º
c温度范围内失重1.5%。
[0133]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其热重分析曲线(tga)图谱基本如图77所示。
[0134]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其差示扫描量热(dsc)曲线在123.7
±
5℃处具有吸热峰。
[0135]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其差示扫描量热图谱基本如图78所示。
[0136]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型b,其动态
水吸附(dvs)图谱基本如图80所示。其dvs曲线在80%rh下的吸湿增重为4.28%
±
0.02%(例如4.28%)。
[0137]
(18)本发明提供了有一种如式i所示的化合物的富马酸盐晶型c,其以2θ角表示的x射线粉末衍射图,在13.557
±
0.2
º
、13.189
±
0.2
º
、16.677
±
0.2
º
、25.518
±
0.2
º
、24.545
±
0.2
º
、25.208
±
0.2
º
、24.137
±
0.2
º
和20.727
±
0.2
º
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:21.208
±
0.2
º
、20
±
0.2
º
、18.603
±
0.2
º
、21.777
±
0.2
º
、17.923
±
0.2
°
、22.991
±
0.2
º
、28.069
±
0.2
º
、11.062
±
0.2
º
、11.46
±
0.2
º
、15.577
±
0.2
º
、15.216
±
0.2
º
、33.735
±
0.2
º
和8.939
±
0.2
º
。
[0138]
在本发明一些实施例方案中,所述的如式i所示的化合物的富马酸盐晶型c,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表22所示。
[0139]
在本发明有一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其xrpd图谱基本如图81所示。
[0140]
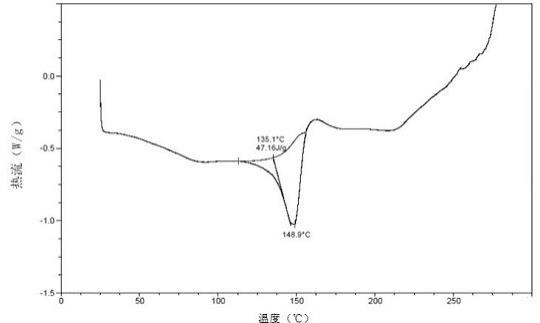
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其热重分析曲线在室温至148.9
º
c温度范围内失重0.78%。
[0141]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其热重分析曲线(tga)图谱基本如图82所示。
[0142]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其差示扫描量热(dsc)曲线在143.71
±
5℃处具有吸热峰。
[0143]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其差示扫描量热图谱基本如图83所示。
[0144]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型c,其动态水吸附(dvs)图谱基本如图85所示。其dvs曲线在80%rh下的吸湿增重为2.04%
±
0.02%(例如2.04%)。
[0145]
(19)本发明提供了有一种如式i所示的化合物的富马酸盐晶型d,其以2θ角表示的x射线粉末衍射图,在13.499
±
0.2
º
、18.016
±
0.2
º
、16.868
±
0.2
º
、17.554
±
0.2
º
、25.983
±
0.2
º
、24.895
±
0.2
º
和11.045
±
0.2
º
处有衍射峰;在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其以2θ角表示的x射线粉末衍射图,还在如下一个或多个2θ角处有衍射峰:25.672
±
0.2
º
、25.187
±
0.2
º
、23.807
±
0.2
º
、21.175
±
0.2
º
、12.052
±
0.2
º
、27.581
±
0.2
º
、11.474
±
0.2
º
、22.382
±
0.2
º
、5.807
±
0.2
º
、18.406
±
0.2
º
、19.95
±
0.2
º
、22.831
±
0.2
º
、16.421
±
0.2
º
、23.43
±
0.2
º
、21.721
±
0.2
º
、26.979
±
0.2
º
、14.475
±
0.2
º
、19.386
±
0.2
º
、20.806
±
0.2
º
、6.996
±
0.2
º
、18.734
±
0.2
º
、20.198
±
0.2
º
、30.874
±
0.2
º
、7.817
±
0.2
º
、30.252
±
0.2
º
、31.238
±
0.2
º
、28.767
±
0.2
º
、19.128
±
0.2
º
、38.281
±
0.2
º
、8.859
±
0.2
º
、34.643
±
0.2
º
、36.76
±
0.2
º
、36.023
±
0.2
º
、15.091
±
0.2
º
、15.45
±
0.2
º
、12.552
±
0.2
º
和10.018
±
0.2
º
。
[0146]
在本发明一些实施例方案中,所述的如式i所示的化合物的富马酸盐晶型d,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表23所示。
[0147]
在本发明有一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其xrpd图谱基本如图87所示。
[0148]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其热重分析曲线在室温至127
º
c温度范围内失重2.31%。
[0149]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其热重分析曲线(tga)图谱基本如图88所示。
[0150]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其差示扫描量热(dsc)曲线在76.8
±
5℃、113.6
±
5℃和143.71
±
5℃处具有吸热峰。
[0151]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其差示扫描量热图谱基本如图89所示。
[0152]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型d,其动态水吸附(dvs)图谱基本如图91所示。其dvs曲线在80%rh下的吸湿增重为3.42%
±
0.02%(例如3.42%)。
[0153]
在本发明一些实施方案中,所述的x射线粉末衍射图均使用cu-kα辐射谱线测得。
[0154]
(20)本发明提供了有一种如式i所示的化合物的富马酸盐晶型g,其以2θ角表示的x射线粉末衍射图,在24.485
±
0.2
º
、16.695
±
0.2
º
、21.351
±
0.2
º
、20.102
±
0.2
º
、24.097
±
0.2
º
、21.641
±
0.2
º
和12.858
±
0.2
º
处有衍射峰。
[0155]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型g时,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:25.01
±
0.2
º
、14.221
±
0.2
º
、19.424
±
0.2
º
、27.061
±
0.2
º
、25.888
±
0.2
º
、20.551
±
0.2
º
、17.397
±
0.2
º
、6.355
±
0.2
º
、7.161
±
0.2
º
、23.438
±
0.2
º
、30.099
±
0.2
º
、28.044
±
0.2
º
、26.321
±
0.2
º
、23.041
±
0.2
º
、8.379
±
0.2
º
、29.08
±
0.2
º
、29.699
±
0.2
º
、15.62
±
0.2
º
、30.602
±
0.2
º
、30.84
±
0.2
º
、37.594
±
0.2
º
、36.834
±
0.2
º
、32.279
±
0.2
º
、39.016
±
0.2
º
、32.671
±
0.2
º
、38.409
±
0.2
º
和34.266
±
0.2
º
。
[0156]
在本发明一些实施例方案中,所述的如式i所示的化合物的富马酸盐晶型g,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表24所示。
[0157]
在本发明有一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型g,其xrpd图谱基本如图92所示。
[0158]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型g,其热重分析曲线在室温至150℃温度范围内失重1.46%。
[0159]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型g,其热重分析曲线图谱基本如图93所示。
[0160]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型g,其差示扫描量热曲线在102.60℃
±
5℃和130.69℃
±
5℃处具有吸热峰。
[0161]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型g,其差示扫描量热图谱基本如图94所示。
[0162]
(21)本发明提供了有一种如式i所示的化合物的富马酸盐晶型j,其以2θ角表示的x射线粉末衍射图,在6.134
±
0.2
º
、14.045
±
0.2
º
、23.884
±
0.2
º
、4.777
±
0.2
º
、6.916
±
0.2
º
、20.611
±
0.2
º
、24.252
±
0.2
º
和19.557
±
0.2
º
处有衍射峰。
[0163]
在本发明一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型j时,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:15.664
±
0.2
º
、
16.633
±
0.2
º
、17.067
±
0.2
º
、13.714
±
0.2
º
、22.499
±
0.2
º
、17.648
±
0.2
º
、18.685
±
0.2
º
、20.269
±
0.2
º
、20.98
±
0.2
º
、25.558
±
0.2
º
、10.151
±
0.2
º
、12.191
±
0.2
º
、12.422
±
0.2
º
、12.822
±
0.2
º
、13.504
±
0.2
º
、30.037
±
0.2
º
、29.591
±
0.2
º
、29.73
±
0.2
º
、28.2
±
0.2
º
和32.8
±
0.2
º
。
[0164]
在本发明一些实施例方案中,所述的如式i所示的化合物的富马酸盐晶型j,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表25所示。
[0165]
在本发明有一些实施方案中,所述的如式i所示的化合物的富马酸盐晶型j,其xrpd图谱基本如图95所示。
[0166]
(22)本发明提供了有一种如式i所示的化合物的甲磺酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在13.56
±
0.2
º
、20.17
±
0.2
º
、21.58
±
0.2
º
、16.76
±
0.2
º
、16.46
±
0.2
º
、13.18
±
0.2
º
、20.97
±
0.2
º
和6.7
±
0.2
º
处具有衍射峰。
[0167]
在本发明一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:25.39
±
0.2
º
、23.78
±
0.2
º
、10.45
±
0.2
º
、14.89
±
0.2
º
、24.48
±
0.2
º
、26.55
±
0.2
º
、30.46
±
0.2
º
、29.87
±
0.2
°
、28.01
±
0.2
º
、18.67
±
0.2
º
、28.63
±
0.2
º
、27.26
±
0.2
º
、22.21
±
0.2
º
、33.21
±
0.2
º
、18.05
±
0.2
º
和32.12
±
0.2
º
。
[0168]
在本发明一些实施例方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表29所示。
[0169]
在本发明有一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其xrpd图谱基本如图96所示。
[0170]
在本发明一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其热重分析曲线在室温至180℃温度范围内失重3.3%。
[0171]
在本发明一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其热重分析曲线图谱基本如图97所示。
[0172]
在本发明一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其差示扫描量热曲线在175.3℃
±
5℃处具有吸热峰。
[0173]
在本发明一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其差示扫描量热图谱基本如图98所示。
[0174]
在本发明一些实施方案中,所述的如式i所示的化合物的甲磺酸盐晶型a,其动态水分吸附图谱基本如图100所示。其dvs曲线在80%rh下的吸湿增重为12.48%
±
0.02%。
[0175]
(23)本发明提供了有一种如式i所示的化合物的草酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在11.95
±
0.2
º
、13.57
±
0.2
º
、17.67
±
0.2
º
、22.76
±
0.2
º
、26.18
±
0.2
°
、20.04
±
0.2
º
、16.76
±
0.2
º
和24.85
±
0.2
º
处具有衍射峰。
[0176]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型a时,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:23.35
±
0.2
º
、15.39
±
0.2
º
、7.7
±
0.2
º
、10.9
±
0.2
º
、9.98
±
0.2
º
、15.05
±
0.2
º
、19.14
±
0.2
º
、12.56
±
0.2
º
、28.49
±
0.2
º
和21.14
±
0.2
º
。
[0177]
在本发明一些实施例方案中,所述的如式i所示的化合物的草酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表31所示。
[0178]
在本发明有一些实施方案中,所述的如式i所示的化合物的草酸盐晶型a,其xrpd图谱基本如图102所示。
[0179]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型a,其热重分析曲线在室温至150℃温度范围内失重6.7%。
[0180]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型a,其热重分析曲线图谱基本如图103所示。
[0181]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型a,其差示扫描量热曲线在85.5
±
5℃、172.5
±
5℃和190.8
±
5℃处具有吸热峰。
[0182]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型a,其差示扫描量热图谱基本如图104所示。
[0183]
(24)本发明提供了有一种如式i所示的化合物的草酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在13.13
±
0.2
º
、19.62
±
0.2
º
、24.47
±
0.2
º
、17
±
0.2
º
、16.32
±
0.2
º
、25.2
±
0.2
º
、10.22
±
0.2
º
和20.87
±
0.2
º
处具有衍射峰。
[0184]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型b时,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:20.47
±
0.2
º
、26.3
±
0.2
º
、6.53
±
0.2
º
、14.21
±
0.2
º
、13.79
±
0.2
º
、28.79
±
0.2
º
、21.67
±
0.2
º
、15.76
±
0.2
º
、30.37
±
0.2
º
、22.78
±
0.2
º
、29.42
±
0.2
º
、17.8
±
0.2
º
和32.08
±
0.2
º
。
[0185]
在本发明一些实施例方案中,所述的如式i所示的化合物的草酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表32所示。
[0186]
在本发明有一些实施方案中,所述的如式i所示的化合物的草酸盐晶型b,其xrpd图谱基本如图106所示。
[0187]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型b,其热重分析曲线在室温至150℃温度范围内失重4.89%。
[0188]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型b,其热重分析曲线图谱基本如图107所示。
[0189]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型b,其差示扫描量热曲线在123.2
±
5℃和182.4
±
5℃处具有吸热峰。
[0190]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型b,其差示扫描量热图谱基本如图108所示。
[0191]
(25)本发明提供了有一种如式i所示的化合物的草酸盐晶型c,其以2θ角表示的x射线粉末衍射图,在11.89
±
0.2
º
、7.65
±
0.2
º
、5.54
±
0.2
º
、19.72
±
0.2
º
、17.43
±
0.2
º
、22.2
±
0.2
º
和10.88
±
0.2
º
处具有衍射峰。
[0192]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型c,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:16.67
±
0.2
º
、21.29
±
0.2
º
、9.81
±
0.2
º
和25.59
±
0.2
º
。
[0193]
在本发明一些实施例方案中,所述的如式i所示的化合物的草酸盐晶型c,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表33所示。
[0194]
在本发明有一些实施方案中,所述的如式i所示的化合物的草酸盐晶型c,其xrpd图谱基本如图110所示。
[0195]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型c,其热重分析曲线在室温至150℃温度范围内失重5.78%。
[0196]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型c,其热重分析曲线图谱基本如图111所示。
[0197]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型c,其差示扫描量热曲线在42.1
±
5℃、179.4
±
5℃和195.8
±
5℃处具有吸热峰。
[0198]
在本发明一些实施方案中,所述的如式i所示的化合物的草酸盐晶型c,其差示扫描量热图谱基本如图112所示。
[0199]
(26)本发明提供了有一种如式i所示的化合物的氢溴酸盐晶型a,其以2θ角表示的x射线粉末衍射图,在13.28
±
0.2
º
、19.99
±
0.2
º
、16.64
±
0.2
º
、20.51
±
0.2
º
、6.89
±
0.2
º
、22.2
±
0.2
º
、21.07
±
0.2
º
和19.03
±
0.2
º
处有衍射峰。
[0200]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:21.74
±
0.2
º
、17.33
±
0.2
º
、25.53
±
0.2
º
、26.74
±
0.2
º
、18.6
±
0.2
º
、14.11
±
0.2
º
、15.9
±
0.2
º
和30.08
±
0.2
º
。
[0201]
在本发明一些实施例方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表34所示。
[0202]
在本发明有一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其xrpd图谱基本如图114所示。
[0203]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其热重分析曲线在室温至150℃温度范围内失重6.3%。
[0204]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其热重分析曲线图谱基本如图115所示。
[0205]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其差示扫描量热曲线在96.5
±
5℃和140.6
±
5℃处具有吸热峰。
[0206]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型a,其差示扫描量热图谱基本如图116所示。
[0207]
(27)本发明提供了有一种如式i所示的化合物的氢溴酸盐晶型b,其以2θ角表示的x射线粉末衍射图,在6.89
±
0.2
º
、19.01
±
0.2
º
、22.13
±
0.2
º
、13.27
±
0.2
º
、21.33
±
0.2
º
、25.63
±
0.2
º
、26.72
±
0.2
º
和18.58
±
0.2
º
处具有衍射峰。
[0208]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:19.71
±
0.2
º
、12.73
±
0.2
º
、14.62
±
0.2
º
、27.84
±
0.2
º
、20.37
±
0.2
º
、16.98
±
0.2
º
、29.48
±
0.2
º
、28.67
±
0.2
°
、10.96
±
0.2
º
、23.28
±
0.2
º
、34.23
±
0.2
º
、11.67
±
0.2
º
和24.04
±
0.2
º
。
[0209]
在本发明一些实施例方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表35所示。
[0210]
在本发明有一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其xrpd图谱基本如图118所示。
[0211]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其热重分析曲线在室温至150℃温度范围内失重8.7%。
[0212]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其热重分析曲线图谱基本如图119所示。
[0213]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其差示扫描量热曲线在131.8
±
5℃、172.7
±
5℃和178.7
±
5℃处具有吸热峰。
[0214]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型b,其差示扫描量热图谱基本如图120所示。
[0215]
(28)本发明提供了有一种如式i所示的化合物的氢溴酸盐晶型c,其以2θ角表示的x射线粉末衍射图,在19.998
±
0.2
º
、13.26
±
0.2
º
、16.639
±
0.2
º
、20.5
±
0.2
º
、21.043
±
0.2
º
、25.5
±
0.2
º
、6.642
±
0.2
º
和17.321
±
0.2
º
处具有衍射峰。
[0216]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其2θ角表示的x射线粉末衍射图,还在如下一个或多个在2θ角处有衍射峰:25.262
±
0.2
º
、22.239
±
0.2
º
、14.219
±
0.2
º
、31.661
±
0.2
º
、19.662
±
0.2
º
、21.76
±
0.2
º
、15.8
±
0.2
º
、26.778
±
0.2
º
、30.043
±
0.2
º
、26.4
±
0.2
º
、25.921
±
0.2
º
、24.78
±
0.2
º
、27.459
±
0.2
º
、28.059
±
0.2
º
、24.379
±
0.2
º
、13.94
±
0.2
º
、24.005
±
0.2
º
、27.8
±
0.2
º
、29.22
±
0.2
º
、29.621
±
0.2
°
、14.817
±
0.2
º
、32.66
±
0.2
º
、16.159
±
0.2
º
、22.916
±
0.2
º
、9.976
±
0.2
º
、28.619
±
0.2
º
、10.501
±
0.2
º
、30.898
±
0.2
º
、30.335
±
0.2
º
、35.684
±
0.2
º
、32.919
±
0.2
º
、17.876
±
0.2
º
、37.34
±
0.2
º
、38.742
±
0.2
º
、33.344
±
0.2
º
、37.66
±
0.2
º
、39.359
±
0.2
º
、15.377
±
0.2
º
、33.62
±
0.2
º
、34.18
±
0.2
º
、10.982
±
0.2
º
、36.211
±
0.2
º
、35.02
±
0.2
º
、34.698
±
0.2
º
、38.38
±
0.2
º
和12.226
±
0.2
º
。
[0217]
在本发明一些实施例方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其以2θ角和峰高百分比表示的x射线粉末衍射图中,其衍射峰和峰高百分比还可如表36所示。
[0218]
在本发明有一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其xrpd图谱基本如图122所示。
[0219]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其热重分析曲线在室温至149℃温度范围内失重3.26%。
[0220]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其热重分析曲线图谱基本如图123所示。
[0221]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其差示扫描量热曲线在50.98
±
5℃和117.20
±
5℃处具有吸热峰。
[0222]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其差示扫描量热图谱基本如图124所示。
[0223]
在本发明一些实施方案中,所述的如式i所示的化合物的氢溴酸盐晶型c,其动态水分吸附图谱基本如图125所示。其dvs曲线在80%rh下的吸湿增重为6.97%
±
0.02%。
[0224]
本发明中,上述各晶型以及无定型化合物的热重分析曲线所得的失重参数中,所述室温的温度一般是指10~30℃,例如25℃。
[0225]
(1)本发明提供了一种如上所述如式i所示化合物的游离碱晶型a的制备方法,其为方案一或方案二;所述方案一,其包括如下步骤:将反溶剂加入到如式i所示化合物与溶剂的溶液中析晶,得到游离碱晶型a即可;其中,所述如式i所示化合物为游离碱无定型,所述的溶剂为
甲醇;所述的反溶剂为水。
[0226]
在本发明实施方案中,所述方案一中,所述甲醇与所述水的体积比为5:8。实验中发现,在采用该方案制备游离碱晶型a的过程中,若水的比例过低,会形成无定型,水的比例过高则形成水合物。
[0227]
在本发明实施方案中,所述方案一中,所述如式i所示化合物与所述溶剂的重量体积比为20~200g/l,例如60g/l。
[0228]
在本发明实施方案中,所述方案一中,所述析晶的温度为10~30℃,例如25℃。
[0229]
所述方案二,其包括以下步骤:将如式i所示化合物与混合溶剂的混悬液进行转晶,得到游离碱晶型a;所述如式i所示化合物为游离碱无定型,所述溶剂为甲醇和水的混合溶剂。
[0230]
在本发明实施方案中,所述方案二中,所述转晶通过搅拌所述混合液实现。例如将所述混合液在50℃下搅拌15天后,离心取沉淀即为所述游离碱晶型a。
[0231]
在本发明实施方案中,所述方案二中,所述转晶的温度为30~55℃,例如50℃。
[0232]
在本发明实施方案中,所述方案二中,所述甲醇与水的体积比为1:(0.8~1.2),例如1:1。
[0233]
在本发明实施方案中,所述方案二中,所述如式i所示化合物与所述溶剂的重量体积比为200~300g/l,例如250g/l。
[0234]
(2)本发明提供了一种如上所述如式i所示化合物的游离碱晶型b的制备方法,其包括以下步骤:将反溶剂加入到如式i化合物与溶剂的溶液中析晶,得到游离碱晶型b即可;其中,所述如式i所示化合物为游离碱无定型,所述的溶剂为乙酸乙酯;所述的反溶剂为甲基四氢呋喃和/或叔丁基甲醚。
[0235]
在本发明实施方案中,所述溶剂与反溶剂的体积比为1:(4~6),例如1:5。
[0236]
在本发明实施方案中,所述析晶的温度为10~30℃,例如25℃。
[0237]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~100g/l,例如50g/l。
[0238]
(3)本发明提供了一种如上所述如式i所示化合物的游离碱晶型c的制备方法,其包括以下步骤:将如式i所示化合物与溶剂的混合液进行转晶即得;所述如式i所示化合物为游离碱无定型,所述溶剂为甲醇和水。
[0239]
在本发明实施方案中,所述甲醇和所述水的体积比为5:(85~96),例如5:95。
[0240]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0241]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0242]
(4)本发明提供了一种如上所述如式i所示化合物的盐酸盐晶型a的制备方法,其包括以下步骤:将盐酸加入到如式i所示化合物与溶剂的混合液,或者如式i所示化合物盐酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型;所述溶剂为异丙醇、丙酮、乙腈、乙酸乙酯、正庚烷、叔丁基甲醚和甲苯中的一种或多种,或者,所述溶剂为“异丙醇和水”或“乙醇和正庚烷”。
[0243]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0244]
在本发明实施方案中,所述溶剂为异丙醇、丙酮、乙腈、正庚烷、叔丁基甲醚或甲
苯,或者,所述溶剂为乙醇和正庚烷,或者所述溶剂为异丙醇和水;当所述溶剂为乙醇和正庚烷时,所述乙醇和正庚烷的体积比为1:(3~5),例如1:4;当所述溶剂为异丙醇和水时,所述异丙醇和水的体积比为1:(0.8~1.2),例如1:1。
[0245]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0246]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~300g/l,例如40g/l或250g/l。
[0247]
在本发明实施方案中,所述盐酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0248]
(5)本发明提供了一种如上所述如式i所示化合物的盐酸盐晶型b的制备方法,其包括以下步骤:将盐酸加入到如式i所示化合物与溶剂的混合液中,或者如式i所示化合物盐酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为醋酸异丙酯。
[0249]
在本发明实施方案中,所述盐酸与所述如式i所示化合物的摩尔比为(1.8~2.2):1,例如2:1。
[0250]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0251]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0252]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~300g/l,例如40g/l。
[0253]
(6)本发明提供了一种如上所述如式i所示化合物的盐酸盐晶型c的制备方法,其包括以下步骤:将盐酸加入到如式i所示化合物与溶剂的混合液中,或者如式i所示化合物盐酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为醋酸异丙酯。
[0254]
在本发明实施方案中,所述盐酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0255]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0256]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0257]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~300g/l,例如40g/l。
[0258]
(7)本发明提供了一种如上所述如式i所示化合物的盐酸盐无定型的制备方法,其包括以下步骤:将盐酸加入到如式i所示化合物与溶剂的混合液中,再加入反溶剂后析出的固体即为所述盐酸盐无定型;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙醇,所述反溶剂为正庚烷。
[0259]
在本发明实施方案中,所述盐酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0260]
(8)本发明提供了一种如上所述如式i所示化合物的硫酸盐晶型a的制备方法,其包括以下步骤:将硫酸加入到如式i所示化合物与溶剂的混合物中,或者将如式i所示化合物硫酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为丙酮和正庚烷。
[0261]
在本发明实施方案中,所述硫酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0262]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0263]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0264]
在本发明实施方案中,所述溶剂中,所述丙酮和所述正庚烷的体积比为1:(0.8~1.2),例如1:1。
[0265]
(9)本发明提供了一种如上所述如式i所示化合物的硫酸盐晶型b的制备方法,其包括以下步骤:将将硫酸加入到如式i所示化合物与溶剂的混合物中,或者如式i所示化合物硫酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为醋酸异丙酯。
[0266]
在本发明实施方案中,所述硫酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0267]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0268]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0269]
(10)本发明提供了一种如上所述如式i所示化合物的硫酸盐晶型c的制备方法,其包括以下步骤:将硫酸加入到如式i所示化合物与溶剂的混合液,或者如式i所示化合物硫酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为丙酮、或者“丙酮和四氢呋喃”、“丙酮和乙酸乙酯”或者“二氯甲烷和乙酸乙酯”。
[0270]
在本发明实施方案中,所述硫酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0271]
在本发明实施方案中,所述转晶的温度为10~40℃,例如25℃。
[0272]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0273]
在本发明实施方案中,所述混合液中所述如式i所示化合物与所述溶剂的重量体积比为100~300g/l,例如250g/l。
[0274]
在本发明实施方案中,所述溶剂为丙酮和乙酸乙酯时,所述丙酮和乙酸乙酯的体积比为2:1。
[0275]
在本发明实施方案中,所述溶剂为二氯甲烷和乙酸乙酯时,所述二氯甲烷和所述乙酸乙酯的体积比为2:1。
[0276]
在本发明实施方案中,所述溶剂为丙酮和四氢呋喃时,所述丙酮和四氢呋喃的体积比为2:1。
[0277]
(11)本发明提供了一种如上所述如式i所示化合物的马来酸盐晶型a的制备方法,其包括以下步骤:将含“马来酸、如式i所示化合物和溶剂”的混合液进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙醇、异丙醇、正庚烷、叔丁基甲醚、甲苯和水中的一种或多种。
[0278]
在本发明实施方案中,所述马来酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0279]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0280]
在本发明实施方案中,所述转晶为将所述混合液依次在10~30℃(例如25℃)、5~50
℃和-10~-20℃条件下搅拌实现。其中,在5~50℃(或者50~5℃)下搅拌时,较佳地循环两次温度。
[0281]
在本发明实施方案中,所述溶剂为乙醇、异丙醇、正庚烷、叔丁基甲醚、甲苯或水;或者,所述溶剂为“乙醇和正庚烷”的混合溶剂,所述混合溶剂中乙醇和正庚烷的体积比例如为1:4;或者,所述溶剂为异丙醇和水的混合溶剂,所述混合溶剂中,所述异丙醇和所述水的体积比例如为1:1。
[0282]
(12)本发明提供了一种如上所述如式i所示化合物的马来酸盐晶型b的制备方法,其包括以下步骤:将“马来酸、如式i所示化合物和溶剂”的混合液,或者所述马来酸盐晶型a与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙酸乙酯和/或醋酸异丙酯。
[0283]
在本发明实施方案中,所述马来酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0284]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0285]
在本发明实施方案中,所述转晶为将所述混合液依次在10~30℃(例如25℃)、5~50℃和-10~-20℃条件下搅拌实现。其中,在5~50℃(或者50~5℃)下搅拌时,较佳地循环两次温度。
[0286]
(13)本发明提供了一种如上所述如式i所示化合物的磷酸盐晶型a的制备方法,其包括以下步骤:将磷酸加入如式i所示化合物与溶剂的混合液,或者如式i所示化合物磷酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为醋酸异丙酯。
[0287]
在本发明实施方案中,所述磷酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0288]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0289]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0290]
(14)本发明提供了一种如上所述如式i所示化合物的磷酸盐晶型b的制备方法,其包括以下步骤:将磷酸加入如式i所示化合物与溶剂的混合液中,或者将如式i所示化合物磷酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为四氢呋喃。
[0291]
在本发明实施方案中,所述磷酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0292]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0293]
在本发明实施方案中,所述转晶为将所述混合液依次在10~30℃(例如25℃)、5~50℃和-10~-20℃条件下搅拌实现。其中,在5~50℃(或者50~5℃)下搅拌时,较佳地循环两次温度。
[0294]
(15)本发明提供了一种如上所述如式i所示化合物的富马酸盐无定型的制备方法,其包括以下步骤:将富马酸、如式i所示化合物和溶剂的混合液中转晶得到;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙醇和正庚烷。
[0295]
在本发明实施方案中,所述富马酸与所述如式i所示化合物的摩尔比为(1~1.2):
1,例如1:1。
[0296]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0297]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0298]
在本发明实施方案中,所述溶剂中,所述乙醇和所述正庚烷的体积比为1:(2~4),例如1:4。
[0299]
(16)本发明提供了一种如上所述如式i所示化合物的富马酸盐晶型a的制备方法,其包括以下步骤:将如式i所示化合物、富马酸和溶剂的混合液,或者将如式i所示化合物富马酸盐的无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型;所述转晶的温度为0~50℃;当所述转晶的温度为0~35℃时,所述溶剂包括异丙醇、丙酮、甲基异丁酮、醋酸异丙酯、甲基叔丁基醚、四氢呋喃、2-甲基四氢呋喃、甲苯、正庚烷和1,4-二氧六烷中的一种或多种;当所述转晶的温度为35~50℃但不包括35℃时,所述溶剂为甲基异丁酮、醋酸异丙酯、甲基叔丁基醚、2-甲基四氢呋喃、甲苯、正庚烷中的一种或多种。
[0300]
在本发明实施方案中,所述富马酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0301]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0302]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0303]
在本发明实施方案中,所述转晶的温度为0~50℃。
[0304]
在本发明实施方案中,当所述转晶的温度为0~35℃时,所述溶剂为异丙醇、丙酮、甲基异丁酮、醋酸异丙酯、甲基叔丁基醚、四氢呋喃、2-甲基四氢呋喃、甲苯、正庚烷或1,4-二氧六烷;或者为丙酮和乙酸乙酯的混合溶剂,所述丙酮与所述乙酸乙酯的体积比例如为2:1;或者为乙醇和正庚烷的混合溶剂,所述乙醇和正庚烷的体积比例如为1:4。
[0305]
(17)本发明提供了一种如上所述如式i所示化合物的富马酸盐晶型b的制备方法,其包括以下步骤:将如式i所示化合物、富马酸和溶剂的混合液,或者将如式i所示化合物的富马酸盐无定型与溶剂的混合液,或者将所述富马酸盐晶型a与溶剂的混合液,依次进行转晶、离心和干燥即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙腈。
[0306]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0307]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0308]
在本发明实施方案中,所述干燥的温度为10~30℃,例如25℃。
[0309]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0310]
(18)本发明提供了一种如上所述如式i所示化合物的富马酸盐晶型c的制备方法,其包括以下步骤:将如式i所示化合物、富马酸和溶剂的混合液,或者将如式i所示化合物的富马酸盐无定型与溶剂的混合液,或者将所述富马酸盐晶型a与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙酸乙酯。
[0311]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0312]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0313]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0314]
(19)本发明提供了一种如上所述如式i所示化合物的富马酸盐晶型d的制备方法,为方案一或方案二:所述方案一包括以下步骤:将如式i所示化合物、富马酸和溶剂的混合液,或者将如式i所示化合物的富马酸盐无定型与溶剂的混合液,或者将所述富马酸盐晶型a与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为水。
[0315]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0316]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0317]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0318]
所述方案二包括以下步骤:将所述富马酸盐晶型a置于在水活度0.8以上的溶剂中转晶,得到富马酸盐晶型d。
[0319]
在本发明实施方案中,水活度0.8以上的溶剂为甲醇和水的混合溶剂。
[0320]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0321]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0322]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0323]
(20)本发明提供了一种如上所述如式i所示化合物的富马酸盐晶型g的制备方法,其包括以下步骤:将如式i所示化合物、富马酸和溶剂的混合液,或者将如式i所示化合物的富马酸盐无定型与溶剂的混合液,或者将所述富马酸盐晶型a与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为甲醇和水。
[0324]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0325]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0326]
在本发明实施方案中,所述甲醇和水的体积比为(80~90):15,例如85:15。
[0327]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0328]
(21)本发明提供了一种如上所述如式i所示化合物的富马酸盐晶型j的制备方法,其包括以下步骤:将如式i所示化合物、富马酸和溶剂的混合液,或者将如式i所示化合物的富马酸盐无定型与溶剂的混合液,或者将所述富马酸盐晶型a与溶剂的混合液,依次进行转晶和离心后,未经干燥的固体即为所述富马酸盐晶型j;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙腈。
[0329]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0330]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0331]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0332]
(22)本发明提供了一种如上所述如式i所示化合物的甲磺酸盐晶型a的制备方法,其包括以下步骤:将如式i所示化合物、甲磺酸和溶剂的混合液,或者将如式i所示化合物的甲磺酸盐无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙醇、乙腈、丙酮、乙酸乙酯、叔丁基甲醚、甲基四氢呋喃、甲苯、二氯甲烷、四氢呋喃和醋酸异丙酯中的一种或多种。
[0333]
在本发明实施方案中,所述甲磺酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0334]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0335]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0336]
在本发明实施方案中,所述如式i所示化合物与所述溶剂的重量体积比为20~60g/l,例如40g/l。
[0337]
(23)本发明提供了一种如上所述如式i所示化合物的草酸盐晶型a的制备方法,其包括以下步骤:将如式i所示化合物、草酸和溶剂的混合液,或者将如式i所示化合物的草酸盐无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为丙酮和正庚烷。
[0338]
在本发明实施方案中,所述草酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0339]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0340]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0341]
在本发明实施方案中,所述丙酮与所述正庚烷的体积比为1:(1~1.2),例如1:1。
[0342]
(24)本发明提供了一种如上所述如式i所示化合物的草酸盐晶型b的制备方法,其包括以下步骤:将如式i所示化合物、草酸和溶剂的混合液,或者将如式i所示化合物的草酸盐无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为醋酸异丙酯。
[0343]
在本发明实施方案中,所述草酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0344]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0345]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0346]
(25)本发明提供了一种如上所述如式i所示化合物的草酸盐晶型c的制备方法,其包括以下步骤:将如式i所示化合物、草酸和溶剂的混合液,或者将如式i所示化合物的草酸盐无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为四氢呋喃。
[0347]
在本发明实施方案中,所述草酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0348]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0349]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0350]
(26)本发明提供了一种如上所述如式i所示化合物的氢溴酸盐晶型a的制备方法,其包括以下步骤:将如式i所示化合物、氢溴酸和混合液,或者将如式i所示化合物的氢溴酸
盐无定型与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为醋酸异丙酯,所述氢溴酸与所述如式i所示化合物的摩尔比为(1~1.2):1。
[0351]
在本发明实施方案中,所述氢溴酸与所述如式i所示化合物的摩尔比为1:1。
[0352]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0353]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0354]
(27)本发明提供了一种如上所述如式i所示化合物的氢溴酸盐晶型b的制备方法,其包括以下步骤:将如式i所示化合物、氢溴酸和混合液,或者将如式i所示化合物的氢溴酸盐无定型与溶剂的混合液,或者将所述氢溴酸盐晶型a与溶剂的混合液,进行转晶即得;其中,所述如式i所示化合物为游离碱无定型,所述氢溴酸与所述如式i所示化合物的摩尔比为(1.8~2.2):1;所述溶剂为醋酸异丙酯、异丙醇、乙酸乙酯、正庚烷、叔丁基甲醚、甲苯中的一种或多种,或者所述溶剂为异丙醇和水。
[0355]
在本发明实施方案中,所述氢溴酸与所述如式i所示化合物的摩尔比为2:1。
[0356]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0357]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0358]
在本发明实施方案中,所述溶剂为醋酸异丙酯、异丙醇、乙酸乙酯、正庚烷、叔丁基甲醚或甲苯。
[0359]
在本发明实施方案中,当所述溶剂为异丙醇和水时,所述异丙醇和水的体积比为1:1。
[0360]
(28)本发明提供了一种如上所述如式i所示化合物的氢溴酸盐晶型c的制备方法,其包括以下步骤:将反溶剂加入到含如式i所示化合物、氢溴酸和溶剂的混合液中析晶即得;其中,所述如式i所示化合物为游离碱无定型,所述溶剂为乙醇,所述反溶剂为正庚烷。
[0361]
在本发明实施方案中,所述氢溴酸与所述如式i所示化合物的摩尔比为(1~1.2):1,例如1:1。
[0362]
在本发明实施方案中,所述转晶通过搅拌所述混合液实现。
[0363]
在本发明实施方案中,所述转晶的温度为10~30℃,例如25℃。
[0364]
在本发明实施方案中,所述乙醇和所述正庚烷的体积比为1:(4~6),例如1:5。
[0365]
本发明还提供了一种药物组合物,其包括治疗有效量的所述的如式i所示化合物或其药学上可接受的盐,以及药学上可接受的载体。
[0366]
本发明还提供了一种所述如式i所示化合物在用于制备药物中的应用,所述药物可为用于制备癌症。其中,所述癌症为肺癌、胰腺癌或结直肠癌;或,所述癌症为kras突变型癌症。
[0367]
本发明中,术语“kras突变型癌症”是指含kras突变的癌症。kras突变型癌症包括但不限于kras突变型肺癌、kras突变型胰腺癌或kras突变型结直肠癌。
[0368]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0369]
本发明所用试剂和原料均市售可得。
[0370]
本发明的积极进步效果在于:发明人经过探索,出人意料地发现本发明的如式i所示化合物的游离碱无定型,通过不同的制备条件能够得到多种药用适用性较强的晶型,有
望能够更显著的抑制癌症或相关疾病。
附图说明
[0371]
图1. 如式i所示化合物的游离碱无定型的xrpd图谱。
[0372]
图2. 如式i所示化合物的游离碱无定型的tga图谱。
[0373]
图3. 如式i所示化合物的游离碱无定型的dsc图谱。
[0374]
图4. 如式i所示化合物的游离碱无定型的dvs图谱。
[0375]
图5. 如式i所示化合物的游离碱无定型dvs测试前后的xrpd对比图谱。
[0376]
图6. 如式i所示化合物的游离碱晶型a的xrpd图谱。
[0377]
图7. 如式i所示化合物的游离碱晶型a的tga图谱。
[0378]
图8. 如式i所示化合物的游离碱晶型a的dcs图谱。
[0379]
图9. 如式i所示化合物的游离碱晶型a的dvs图谱。
[0380]
图10. 如式i所示化合物的游离碱晶型a dvs测试前后的xrpd对比图谱。
[0381]
图11. 如式i所示化合物的游离碱晶型b的xrpd图谱。
[0382]
图12. 如式i所示化合物的游离碱晶型b的tga图谱。
[0383]
图13. 如式i所示化合物的游离碱晶型b的dsc图谱。
[0384]
图14. 如式i所示化合物的游离碱晶型c的xrpd图谱。
[0385]
图15. 如式i所示化合物的游离碱晶型c的tga图谱。
[0386]
图16. 如式i所示化合物的游离碱晶型c的dsc图谱。
[0387]
图17. 如式i所示化合物的盐酸盐晶型a的xrpd图谱。
[0388]
图18. 如式i所示化合物的盐酸盐晶型a的tga图谱。
[0389]
图19. 如式i所示化合物的盐酸盐晶型a的dsc图谱。
[0390]
图20. 如式i所示化合物的盐酸盐晶型a的h-nmr图谱。
[0391]
图21. 如式i所示化合物的盐酸盐晶型a的dvs图谱。
[0392]
图22. 如式i所示化合物的盐酸盐晶型b的xrpd图谱。
[0393]
图23. 如式i所示化合物的盐酸盐晶型b的tga图谱。
[0394]
图24. 如式i所示化合物的盐酸盐晶型b的dsc图谱。
[0395]
图25. 如式i所示化合物的盐酸盐晶型b的1h-nmr图谱。
[0396]
图26. 如式i所示化合物的盐酸盐晶型c的xrpd图谱。
[0397]
图27. 如式i所示化合物的盐酸盐晶型c的tga图谱。
[0398]
图28. 如式i所示化合物的盐酸盐晶型c的dsc图谱。
[0399]
图29. 如式i所示化合物的盐酸盐晶型c的1h-nmr图谱。
[0400]
图30. 如式i所示化合物的盐酸盐无定型的xrpd图谱。
[0401]
图31. 如式i所示化合物的盐酸盐无定型的tga图谱。
[0402]
图32. 如式i所示化合物的盐酸盐无定型的dsc图谱。
[0403]
图33. 如式i所示化合物的硫酸盐晶型a的xrpd图谱。
[0404]
图34. 如式i所示化合物的硫酸盐晶型a的tga图谱。
[0405]
图35. 如式i所示化合物的硫酸盐晶型a的dsc图谱。
[0406]
图36. 如式i所示化合物的硫酸盐晶型a的1h-nmr图谱。
[0407]
图37. 如式i所示化合物的硫酸盐晶型a的dvs图谱。
[0408]
图38. 如式i所示化合物的硫酸盐晶型a dvs测试前后的xrpd对比图谱。
[0409]
图39. 如式i所示化合物的硫酸盐晶型b的xrpd图谱。
[0410]
图40. 如式i所示化合物的硫酸盐晶型b的tga图谱。
[0411]
图41. 如式i所示化合物的硫酸盐晶型b的dsc图谱。
[0412]
图42. 如式i所示化合物的硫酸盐晶型b的1h-nmr图谱。
[0413]
图43. 如式i所示化合物的硫酸盐晶型c的xrpd图谱。
[0414]
图44. 如式i所示化合物的硫酸盐晶型c的tga图谱。
[0415]
图45. 如式i所示化合物的硫酸盐晶型c的dsc图谱。
[0416]
图46. 如式i所示化合物的硫酸盐晶型c的1h-nmr图谱。
[0417]
图47. 如式i所示化合物的硫酸盐晶型c的dvs图谱。
[0418]
图48. 如式i所示化合物的硫酸盐晶型c的dvs测试前后的xrpd对比图谱。
[0419]
图49. 如式i所示化合物的马来酸盐晶型a的xrpd图谱。
[0420]
图50. 如式i所示化合物的马来酸盐晶型a的tga图谱。
[0421]
图51. 如式i所示化合物的马来酸盐晶型a的dsc图谱。
[0422]
图52. 如式i所示化合物的马来酸盐晶型a的1h-nmr图谱。
[0423]
图53. 如式i所示化合物的马来酸盐晶型a的dvs图谱。
[0424]
图54. 如式i所示化合物的马来酸盐晶型b的xrpd图谱。
[0425]
图55. 如式i所示化合物的马来酸盐晶型b的tga图谱。
[0426]
图56. 如式i所示化合物的马来酸盐晶型b的dsc图谱。
[0427]
图57. 如式i所示化合物的马来酸盐晶型b的1h-nmr图谱。
[0428]
图58. 如式i所示化合物的磷酸盐晶型a的xrpd图谱。
[0429]
图59. 如式i所示化合物的磷酸盐晶型a的tga图谱。
[0430]
图60. 如式i所示化合物的磷酸盐晶型a的dsc图谱。
[0431]
图61. 如式i所示化合物的磷酸盐晶型a的1h-nmr图谱。
[0432]
图62. 如式i所示化合物的磷酸盐晶型b的xrpd图谱。
[0433]
图63. 如式i所示化合物的磷酸盐晶型b的tga图谱。
[0434]
图64. 如式i所示化合物的磷酸盐晶型b的dsc图谱。
[0435]
图65. 如式i所示化合物的磷酸盐晶型b的1h-nmr图谱。
[0436]
图66. 如式i所示化合物的富马酸盐无定型的xrpd图谱。
[0437]
图67. 如式i所示化合物的富马酸盐无定型的tga图谱。
[0438]
图68. 如式i所示化合物的富马酸盐无定型的dsc图谱。
[0439]
图69. 如式i所示化合物的富马酸盐无定型的1h-nmr图谱。
[0440]
图70. 如式i所示化合物的富马酸盐晶型a的xrpd图谱。
[0441]
图71. 如式i所示化合物的富马酸盐晶型a的tga图谱。
[0442]
图72. 如式i所示化合物的富马酸盐晶型a的dsc图谱。
[0443]
图73. 如式i所示化合物的富马酸盐晶型a的1h-nmr图谱。
[0444]
图74. 如式i所示化合物的富马酸盐晶型a的dvs图谱。
[0445]
图75. 如式i所示化合物的富马酸盐晶型a dvs测试前后的xrpd对比图谱。
[0446]
图76. 如式i所示化合物的富马酸盐晶型b的xrpd图谱。
[0447]
图77. 如式i所示化合物的富马酸盐晶型b的tga图谱。
[0448]
图78. 如式i所示化合物的富马酸盐晶型b的dsc图谱。
[0449]
图79. 如式i所示化合物的富马酸盐晶型b的1h-nmr图谱。
[0450]
图80. 如式i所示化合物的富马酸盐晶型b的dvs图谱。
[0451]
图81. 如式i所示化合物的富马酸盐晶型c的xrpd图谱。
[0452]
图82. 如式i所示化合物的富马酸盐晶型c的tga图谱。
[0453]
图83. 如式i所示化合物的富马酸盐晶型c的dsc图谱。
[0454]
图84. 如式i所示化合物的富马酸盐晶型c的1h-nmr图谱。
[0455]
图85. 如式i所示化合物的富马酸盐晶型c的dvs图谱。
[0456]
图86. 如式i所示化合物的富马酸盐晶型c dvs测试前后的xrpd对比图谱。
[0457]
图87. 如式i所示化合物的富马酸盐晶型d的xrpd图谱。
[0458]
图88. 如式i所示化合物的富马酸盐晶型d的tga图谱。
[0459]
图89. 如式i所示化合物的富马酸盐晶型d的dsc图谱。
[0460]
图90. 如式i所示化合物的富马酸盐晶型d的1h-nmr谱。
[0461]
图91. 如式i所示化合物的富马酸盐晶型d的dvs图谱。
[0462]
图92. 如式i所示化合物的富马酸盐晶型g的xrpd图谱。
[0463]
图93. 如式i所示化合物的富马酸盐晶型g的tga图谱。
[0464]
图94. 如式i所示化合物的富马酸盐晶型g的dsc图谱。
[0465]
图95. 如式i所示化合物的富马酸盐晶型j的xrpd图谱。
[0466]
图96. 如式i所示化合物的甲磺酸盐晶型a的xrpd图谱。
[0467]
图97. 如式i所示化合物的甲磺酸盐晶型a的tga图谱。
[0468]
图98. 如式i所示化合物的甲磺酸盐晶型a的dsc图谱。
[0469]
图99. 如式i所示化合物的甲磺酸盐晶型a的1h-nmr图谱。
[0470]
图100. 如式i所示化合物的甲磺酸盐晶型a的dvs图谱。
[0471]
图101. 如式i所示化合物的甲磺酸盐晶型a的dvs测试前后的xrpd对比图谱。
[0472]
图102. 如式i所示化合物的草酸盐晶型a的xrpd图谱。
[0473]
图103. 如式i所示化合物的草酸盐晶型a的tga图谱。
[0474]
图104. 如式i所示化合物的草酸盐晶型a的dsc图谱。
[0475]
图105. 如式i所示化合物的草酸盐晶型a的1h-nmr图谱。
[0476]
图106. 如式i所示化合物的草酸盐晶型b的xrpd图谱。
[0477]
图107. 如式i所示化合物的草酸盐晶型b的tga图谱。
[0478]
图108. 如式i所示化合物的草酸盐晶型b的dsc图谱。
[0479]
图109. 如式i所示化合物的草酸盐晶型b的1h-nmr图谱。
[0480]
图110. 如式i所示化合物的草酸盐晶型c的xrpd图谱。
[0481]
图111. 如式i所示化合物的草酸盐晶型c的tga图谱。
[0482]
图112. 如式i所示化合物的草酸盐晶型c的dsc图谱。
[0483]
图113. 如式i所示化合物的草酸盐晶型c的1h-nmr图谱。
[0484]
图114. 如式i所示化合物的氢溴酸盐晶型a的xrpd图谱。
[0485]
图115. 如式i所示化合物的氢溴酸盐晶型a的tga图谱。
[0486]
图116. 如式i所示化合物的氢溴酸盐晶型a的dsc图谱。
[0487]
图117. 如式i所示化合物的氢溴酸盐晶型a的1h-nmr图谱。
[0488]
图118. 如式i所示化合物的氢溴酸盐晶型b的xrpd图谱。
[0489]
图119. 如式i所示化合物的氢溴酸盐晶型b的tga图谱。
[0490]
图120. 如式i所示化合物的氢溴酸盐晶型b的dsc图谱。
[0491]
图121. 如式i所示化合物的氢溴酸盐晶型b的1h-nmr图谱。
[0492]
图122. 如式i所示化合物的氢溴酸盐晶型c的xrpd图谱。
[0493]
图123. 如式i所示化合物的氢溴酸盐晶型c的tga图谱。
[0494]
图124. 如式i所示化合物的氢溴酸盐晶型c的dsc图谱。
[0495]
图125. 如式i所示化合物的氢溴酸盐晶型c的dvs图谱。
具体实施方式
[0496]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0497]
在本发明中,温度以摄氏度(℃)表示,操作温度在室温环境下进行,所示室温是指10~30℃,一般指25℃;所述熔点的可允许误差在
±
1%。
[0498]
本发明所使用的所有溶剂是市售的,无需进一步纯化即可使用。
[0499]
射线粉末衍射(x-ray powder diffractometer, xrpd)实施例样品的xrpd数据由bruker公司 d8 advance x-射线衍射仪测定,检测参数如下:x-ray发生器:cu, kα, (λ=1.54056
ǻ
).光管电压:40 kv,光管电流:40 ma.扫描范围(2θ角):4-40
°
步长:0.02
°
速率:0.1秒/步差式扫描量热分析(differential scanning calorimeter, dsc)实施例样品的dsc数据由ta公司q2000差示扫描量热仪测定,检测参数如下:温度范围(
°
c):25
‑ꢀ
300升温速率(
°
c /min):10保护气:氮气保护气流量(ml /min):50热重分析(thermal gravimetric analyzer, tga)实施例样品的tga数据由ta公司q5000热重分析仪测定,检测参数如下:温度范围(
°
c):25-300升温速率(
°
c /min):10保护气:氮气保护气流量(ml /min):60
动态蒸汽吸附分析(dynamic vapor sorption, dvs)实施例样品的dvs数据由sms公司的 dvs advantage动态蒸汽吸附仪测定,检测参数如下:温度:25
°
c平衡:dm/dt=0.01 %/min(最长平衡时间120 min)干燥:0% rh下干燥5minrh(%)测试梯级:5%rh(%)测试梯级范围:0%
ꢀ‑ꢀ
95%-0%吸湿性评价分类标准如下表h所示:注:δw%表示受试品在25
ꢀ±ꢀ1°
c和80
ꢀ±ꢀ
2% rh下的吸湿增重。
[0500]
高效液相色谱分析方法(high performance liquid chromatograph, hplc)溶解度实验和固态稳定性实验的hplc检测方法参见下表g:
实施例11 游离碱无定型通过参照如专利号cn113087700a中实施例49的方法来制备如式i化合物(专利中化合物45)合成2-((s)-4-((r)-4-氯-2
’‑
((四氢-1h-吡咯嗪-7a(5h)-基)甲氧基)-2,3,5’,8
’‑
四氢-6’h-螺[茚-1,7
’‑
喹唑啉]-4
’‑
基)哌嗪-2-基)乙腈在ar下于0℃下向2-((s)-4-((r)-4-氯-2
’‑
((四氢-1h-吡咯嗪-7a(5h)-基)甲氧基)-2,3,5’,8
’‑
四氢-6’h-螺[茚-1,7
’‑
喹唑啉]-4
’‑
基)哌嗪-2-基)乙腈(1.0g,1.88mmol)、dmap (0.23g,1.88mmol)、tea即三乙胺(0.57g,5.63mmol)和2-氟丙烯酸(0.51g,5.63mmol)在二氯甲烷(简称dcm,20ml)中的溶液中加入t3p即丙基磷酸酐(2.39g,3.75mmol),然后将混合物于室温下搅拌1小时。加入水,将所得混合物用dcm萃取三次。合并有机层,用盐水洗涤,用na2so4干燥,过滤并真空浓缩,得到残余物,其通过制备-hplc纯化,得到呈白色固体的如式i化合物(410mg,36.1%)。
[0501]
ms:605.3[m h
]。
[0502]1h nmr(400mhz,dmso) δ7.30-7.18(m,2h),7.14(dd,j=7.0,1.2hz,1h),5.39(dd,j=18.0,4.1hz,1h),5.35
–
5.15(m,1h),5.08
–
4.40(m,1h),4.32-3.61(m,6h),3.34-3.09(m,2h),3.05
–
2.81(m,6h),2.80
–
2.54(m,6h),2.11
–
2.03(m,1h),1.98
–
1.65(m,9h),1.63-1.49(m,2h)。
[0503]
上述方法中制备得到的呈白色固体的如式i化合物的检测如下:xrpd表征如图1所示。由图可知,采用上述制备方法制得的如式i所示化合物为游离碱无定型。
[0504]
tga结果(图2)显示样品在165.4℃前失重0.09%。
[0505]
dsc结果(图3)显示样品的玻璃化转化温度为81.50℃(中间值)。
[0506]
dvs结果(图4和图5)显示游离碱在80%rh下吸湿增重8.62%,有引湿性,吸湿前后晶型未发生变化。
[0507]
2 游离碱晶型a将实施例1中方法制得的如式i化合物300mg(游离碱,无定型)加入到40ml玻璃瓶中,然后加入5ml甲醇,待样品全溶后,缓慢滴加水(~8ml),待析出固体后,继续室温搅拌2天,离心去除上清液,所得沉淀真空(25℃,-0.1mpa)干燥。
[0508]
xrpd结果如图6所示,衍射数据如下表1。
[0509]
tga结果(图7)显示游离碱晶型a在148.3℃之前失重0.65%。
[0510]
dsc结果(图8)显示样品在121.63℃(峰值温度)有一个吸热峰。
[0511]
dvs结果(图9-图10)显示样品在80%rh吸湿增重1.82%,略有引湿性,吸湿前后晶型未发生变化。
[0512]
表. 游离碱晶型a的xrpd衍射数据
3 游离碱晶型b将实施例1中方法制得的如式i化合物30mg(游离碱,无定型)加入到8ml玻璃瓶中,然后加入0.6 ml乙酸乙酯(简称etoac),待样品全溶后,缓慢滴加甲基四氢呋喃(3 ml),待析出固体后,继续室温搅拌2天,离心去除上清液,所得沉淀真空(25℃,-0.1mpa)干燥。
[0513]
xrpd表征结果如图11所示,xrpd衍射数据如下表2所示。
[0514]
tga结果(图12)显示样品在室温加热到139℃后失重3.96%。
[0515]
dsc结果(图13)显示样品在111.30℃(峰值温度)有一个吸热峰。
[0516]
4 游离碱晶型c游离碱晶型c为水合物晶型,由实施例1中方法制得的如式i化合物300mg(游离碱,无定型)于室温下,在5ml的混合溶剂(meoh-h2o=5/95)混悬打浆制备得到。
[0517]
xrpd表征结果如图14所示,衍射数据如下表3。
[0518]
tga结果(图15)显示样品从室温加热到133.1℃后失重6.75%。
[0519]
dsc结果(图16)显示样品在70.59℃和124.14℃(峰值温度)有两个吸热峰。
[0520]
5 游离碱多晶型的筛选试验1、悬浮液法取50mg的如式i化合物(采用实施例1的制备方法制得的如式i化合物)到2ml的玻璃瓶中,加入搅拌子,然后分别添加200μl溶剂(如下表所示),所得混悬液在25℃或50℃下搅拌15天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表4。
[0521]
2. 反溶剂法取150 mg的如式i化合物(采用实施例1的制备方法制得的如式i化合物)到8 ml的玻璃瓶中,然后分别添加3 ml溶剂,得到的溶液经过0.22 μm的尼龙滤膜过滤后,所得滤液均分成5份,加入到8 ml玻璃瓶中,每份约0.6 ml,边搅拌(600 rpm)边向该澄清溶液中滴加反溶剂(如下表5所示),直至有固体析出,或当反溶剂总体积加至3ml后,若仍无固体析出,不再加入溶剂,所有混悬样品在室温下搅拌3天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥并检测。其结果参见表5。
[0522]
3. 挥发法采用不同的溶剂体系共设置了10个缓慢挥发试验。分别称取约30mg的化合物至4ml小瓶中,使用0.22μm的尼龙滤头过滤,用parafilm
®
封口膜封住小瓶,并在上面扎有针孔,放置在室温下缓慢挥发。收集所得固体并进行xrpd 测试,其结果参见表6。
[0523]
发明人在研发过程中发现游离碱无定型在特定的不同溶剂中能够形成不同的晶型,其中,游离碱无定型在甲醇和水的溶剂中,以不同的配比能够形成游离碱晶型a和游离碱晶型c,而在乙酸乙酯和甲基四氢呋喃中则形成了游离碱晶型b。经检测,三种晶型中游离碱晶型a的引湿性最低,且热稳定性好,同时,根据其在不同生物溶媒中的溶解性实验可知,其在sgf和fessif v1(ph 5.0)中具有较佳地溶解度,由此可见,游离碱晶型a在制备成药物制剂时,相对于无定型以及其他游离碱晶型具有更高的生物利用度。
[0524]
下述提到的酸碱摩尔比是指游离碱起始样品与酸(例如盐酸、硫酸或富马酸)的摩尔比。
[0525]
实施例2 盐酸盐晶型的制备1 盐酸盐晶型a盐酸盐晶型a由游离碱无定型(由实施例1中方法制得的如式i化合物)溶于乙醇(简称etoh)/正庚烷 (1:4,v/v,重量体积比为40g/l)后,加入盐酸(酸碱投料摩尔比1:1),在室温下搅拌3天后,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0526]
xrpd表征结果如图17所示,衍射数据如下表7所示。
[0527]
tga结果(图18)显示样品在室温加热到160℃之前有8.93%的失重。
[0528]
dsc结果(图19)显示样品在158.4℃(峰值温度)有1个吸热峰。
[0529]1h-nmr结果(图20)显示样品中残留etoh与api的摩尔比为0.03(对应失重0.2%)。
[0530]
uplc/ic 结果显示游离碱与盐酸的摩尔比为1:1.8。
[0531]
dvs结果(图21)显示样品在80%rh下吸湿5.02%,有引湿性。
[0532]
2 盐酸盐晶型b盐酸盐晶型b由游离碱无定型(由实施例1中方法制得的如式i化合物)与醋酸异丙酯(简称ipac)(重量体积比为40g/l)混合后,加入盐酸(酸碱投料摩尔比2:1),并在室温下搅拌约3天后,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0533]
xrpd表征结果如图22所示,衍射数据如下表8所示。
[0534]
tga结果(图23)显示晶型b从室温加热至160℃时有12.68% 的失重。
[0535]
dsc结果(图24)显示晶型b在153.2和169.8℃峰值温度)有2个吸热峰。
[0536]1h-nmr 结果(图25)显示未检测到ipac溶剂残留。
[0537]
uplc/ic 结果显示样品酸碱摩尔比为2.4。
[0538]
3 盐酸盐晶型c盐酸盐晶型c由游离碱无定型(由实施例1中方法制得的如式i化合物)与ipac(重量体积比为40g/l)混合后,加入盐酸(酸碱投料摩尔比1:1),并在室温下搅拌约3天后,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0539]
xrpd表征结果如图26所示,衍射数据如下表9所示。
[0540]
tga结果(图27)显示样品从室温加热至150
º
c时有7.82%的失重。
[0541]
dsc结果(图28)显示样品在105.6、131.5和140.9
º
c(峰值温度)有3个吸热峰。
[0542]1h-nmr结果(图29)显示样品中残留ipac与api的摩尔比为0.03(对应失重0.4%)。
[0543]
uplc/ic结果显示样品游离碱与盐酸的摩尔比为1:1.5。
[0544]
4 盐酸盐无定型盐酸盐无定型由游离碱无定型(由实施例1中方法制得的如式i化合物)与乙醇混
合后,加入盐酸(酸碱投料摩尔比1:1),然后滴加反溶剂正庚烷,析出固体后在室温下搅拌约3天后,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0545]
xrpd表征结果如图30所示。
[0546]
tga结果(图31)显示样品从室温加热至131℃时有3.4%的失重。
[0547]
dsc结果(图32)显示样品的玻璃化转化温度是101.66℃。
[0548]
5 盐酸盐多晶型筛选试验取50mg上述制得的盐酸盐无定型到2ml的玻璃瓶中,加入搅拌子,然后分别添加200μl溶剂(如下表所示),所得混悬液在25℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表10。
[0549]
发明人在研发过程中发现游离碱无定型在特定的不同溶剂或是不同的酸碱投料比下与盐酸的混合液能够形成不同的盐酸盐晶型,其中,三种晶型中盐酸盐晶型a的引湿性最低,且热稳定性好,同时,根据其在不同生物溶媒中的溶解性实验可知,其在sgf、fessif v1(ph 5.0)和水中具有较佳地溶解度。可见,盐酸盐晶型a在制备成药物制剂时,相对于无定型以及其他盐酸盐晶型具有更高的生物利用度。
[0550]
实施例3 硫酸盐晶型的制备1 硫酸盐晶型a硫酸盐晶型a由游离碱无定型(由实施例1中方法制得的如式i化合物)与丙酮/正庚烷 (1:1,v/v)混合后,加入硫酸(酸碱投料摩尔比1:1)在室温下搅拌约3天后,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0551]
硫酸盐晶型a 的xrpd结果如图33所示,衍射数据如下表11所示。
[0552]
tga结果(图34)显示样品从室温加热至150℃时有6.52%的失重。
[0553]
dsc结果(图35)显示在108.7 和148.3℃(峰值温度)有2个吸热峰。
[0554]1h-nmr结果(图36)显示样品中残留正庚烷与api的摩尔比为0.05(对应失重0.7%)。
[0555]
uplc/ic结果显示游离碱和酸摩尔比为1:1.3。
[0556]
dvs结果(图37-图38)显示样品在80%rh下吸湿增重11.01%,有引湿性,吸湿前后晶型未发生变化。
[0557]
2 硫酸盐晶型b硫酸盐晶型b由游离碱无定型(由实施例1中方法制得的如式i化合物)与ipac混合后,加入硫酸(酸碱投料摩尔比1:1)在室温下搅拌约3天后,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0558]
硫酸盐晶型b的xrpd结果如图39所示,衍射数据如下表12所示。
[0559]
tga结果(图40)显示样品从室温加热至150℃时有7.93%的失重。
[0560]
dsc结果(图41)显示样品在96.5和142.9℃(峰值温度)有2个吸热峰。
[0561]1h-nmr结果(图42)显示样品中残留ipac与api的摩尔比为0.02 (对应失重0.3%)。
[0562]
uplc/ic结果显示游离碱和酸摩尔比为1:1.2。
[0563]
3 硫酸盐晶型c50 mg的采用下述方法制得的硫酸盐无定型化合物到2ml的玻璃瓶中,加入搅拌子,然后添加200μl丙酮,所得混悬液在40℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1 mpa,25℃)干燥即得硫酸盐晶型c。
[0564]
硫酸盐晶型c的xrpd结果如图43所示,衍射数据如下表13所示。
[0565]
tga结果(图44)显示样品加热至150℃时有5.9%的失重。
[0566]
dsc结果(图45)显示样品在148.5℃(峰值温度)有1个吸热峰。
[0567]1h-nmr结果(图46)显示样品未检测到四氢呋喃(简称thf)溶剂残留。
[0568]
uplc/ic 结果显示游离碱和酸的摩尔比为1:1.2。
[0569]
dvs结果(图47-图48)显示样品在80%rh下吸湿增重3.72%,有引湿性。吸湿前后晶型未发生变化。
[0570]
4 硫酸盐无定型称量1g的游离碱起始样品(由实施例1中方法制得的如式i化合物)到40ml玻璃瓶中,加入25ml的thf,得到澄清溶液,缓慢滴加162mg的浓硫酸,有少量固体析出,搅拌2小时,溶液变成澄清液,室温下继续搅拌3天。加入10ml反溶剂正庚烷,得到固体,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0571]
5 硫酸盐多晶型筛选试验悬浮液法取50mg的如式i所示化合物的硫酸盐无定型到2ml的玻璃瓶中,加入搅拌子,然后分别添加200μl溶剂(如下表14所示),所得混悬液在40℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表14。
[0572]
发明人在研发过程中发现游离碱无定型或是硫酸盐无定型在特定的不同溶剂能够形成不同的硫酸盐晶型,三种硫酸盐晶型中硫酸盐晶型c的引湿性最低,且热稳定性好,而硫酸盐晶型a虽然引湿性较高,但是其生物溶解度和固体稳定性却优于硫酸盐晶型c。硫酸盐晶型a和硫酸盐晶型c制备成制剂时各有优势。
[0573]
实施例4 马来酸盐晶型的制备1 马来酸盐晶型a马来酸盐晶型a由游离碱无定型(由实施例1中方法制得的如式i化合物)与马来酸(酸碱投料摩尔比1:1)在etoh/正庚烷(1:4,v/v)室温下搅拌约3天后,在温度循环下(50
º
c~5
º
c,0.1
º
c/min,循环两次温度)搅拌约1天,再转移至-20℃搅拌2天后得到。
[0574]
马来酸盐晶型a的xrpd结果如图49所示,衍射数据如下表15所示。
[0575]
tga结果(图50)显示样品加热至130℃时有9.1%的失重。
[0576]
dsc结果(图51)显示样品在95.0℃(峰值温度)有1个吸热峰。
[0577]1h-nmr结果(图52)显示,样品中马来酸与游离碱摩尔比为1.0,残留溶剂etoh与游离碱摩尔比为0.04(对应失重0.3%),残留溶剂正庚烷与游离碱摩尔比为0.03 (对应失重0.4%)。
[0578]
dvs结果(图53)显示在80%rh下吸湿增重5.93%,有引湿性。
[0579]
2. 马来酸盐晶型b马来酸盐晶型b是由游离碱无定型(由实施例1中方法制得的如式i化合物)与马来酸(酸碱投料摩尔比1:1)在ipac 中室温下搅拌约3天,温度循环下(50℃~5℃,0.1
º
c/min,循环两次温度)搅拌约1天,再转移至-20℃搅拌2天,离心去除上清液后,得到的固体在室温下干燥过夜即得。
[0580]
马来酸盐晶型b的xrpd结果如图(图54)所示,衍射数据如下表16所示。
[0581]
tga 结果(图55)显示样品加热至130℃时有5.0%的失重。
[0582]
dsc结果(图56)显示在92.4和126.7℃(峰值温度)有2个吸热峰。
[0583]1h-nmr结果(图57)显示,样品中马来酸与游离碱摩尔比为1.0,残留溶剂ipac与游离碱摩尔比为0.1 (对应失重1.7%)。
[0584]
3. 马来酸盐多晶型筛选试验悬浮液法取50mg上述制得的马来酸盐晶型a到2ml的玻璃瓶中,加入搅拌子,然后分别添加
200μl溶剂(如下表所示),所得混悬液在25℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表17。
[0585]
发明人在研发过程中发现游离碱无定型在特定的不同溶剂中与马来酸的混合液能够形成不同的马来酸盐晶型,制得的两种马来酸盐晶型中,马来酸盐晶型a的引湿性低,同时在不同生物溶媒中的溶解性也较高,在制备成药物制剂时,具有较高的生物利用度。
[0586]
实施例5 磷酸盐晶型的制备1. 磷酸盐晶型a磷酸盐晶型a由游离碱无定型(由实施例1中方法制得的如式i化合物)与ipac混合后,加入磷酸(酸碱投料摩尔比1:1)在室温下搅拌约3天后,离心去除上清液,所得的固体干燥后即为磷酸盐晶型a。
[0587]
xrpd结果如图58所示,衍射数据如表18所示。
[0588]
tga结果(图59)显示样品加热至180℃时有6.0% 的失重。
[0589]
dsc结果(图60)显示样品在113.5和161.7℃(峰值温度)有2个吸热峰。
[0590]1h-nmr 结果(图61)显示样品中残留ipac与游离碱摩尔比为0.1(对应失重1.7%)。
[0591]
uplc/ic 结果显示游离碱和磷酸的摩尔比为1:1.5。
[0592]
2. 磷酸盐晶型b磷酸盐晶型b由游离碱无定型(由实施例1中方法制得的如式i化合物)与thf混合后,加入磷酸(酸碱投料摩尔比1:1)后在室温下搅拌约3天,温度循环下(50℃~5℃,0.1
º
c/min,2循环)搅拌约1天,再转移至-20℃搅拌2天后,离心去除上清液,所得的固体干燥后即为磷酸盐晶型b。
[0593]
磷酸盐晶型b的xrpd结果如图62所示,衍射数据如表19所示。
[0594]
tga结果(图63)显示样品加热至90℃时样品有1.4%的失重,90至160℃样品有2.7%的失重。
[0595]
dsc结果(图64)显示样品在77.2和184.0℃(峰值温度)有2个吸热峰。
[0596]1h-nmr结果(图65)显示样品中残留thf与api的摩尔比为0.6(对应失重4.9%)。
[0597]
uplc/ic结果显示样品游离碱和磷酸的摩尔比为1:3.1,可能含有残留的磷酸。
[0598]
实施例6 富马酸盐晶型的制备1 富马酸盐无定型
富马酸盐无定型可由游离碱无定型(由实施例1中方法制得的如式i化合物)与富马酸(酸碱投料摩尔比1:1)在etoh/正庚烷 (1:4,v/v)中室温下搅拌约3天后,离心去除上清液后,所得的固体干燥后即为富马酸盐无定型。
[0599]
富马酸盐无定型xrpd结果如图66所示。
[0600]
tga结果(图67)显示样品加热至163℃时有6.0%的失重。
[0601]
dsc结果(图68)显示在53.5和101.4℃(峰值温度)有1个吸热峰。
[0602]1h nmr结果(图69)显示,样品中富马酸与游离碱摩尔比为1.0,残留溶剂etoh与游离碱摩尔比为0.54(对应失重2.7%),残留溶剂正庚烷 与游离碱摩尔比为0.2(对应失重2.0%)。
[0603]
2. 富马酸盐晶型a富马酸盐晶型a可由1g的游离碱无定型(由实施例1中方法制得的如式i化合物)与富马酸(酸碱投料摩尔比1:1)在25ml的丙酮/乙酸乙酯=2/1(或者乙醇/正庚烷=1/4,v/v)中室温下搅拌约3天后,离心去除上清液后,所得的固体干燥后即为富马酸盐晶型a。
[0604]
富马酸盐晶型a的xrpd结果如图70,衍射数据如下表20所示。
[0605]
tga结果(图71)显示样品加热至150℃时有2.65%的失重。
[0606]
dsc结果(图72)显示在153.1℃(峰值温度)有1个吸热峰。
[0607]1h-nmr 结果(图73)显示样品中游离碱与富马酸的摩尔比为1:1.1,残留溶剂etoh与游离碱摩尔比为0.02(对应失重0.1%),残留溶剂正庚烷与游离碱摩尔比为0.01(对应失重0.1%)。
[0608]
dvs结果(图74-图75)显示样品在80%rh下增重1.41%,略有引湿性,吸湿前后晶型未发生变化。
[0609]
3. 富马酸盐晶型b富马酸盐晶型b可由上述方法制得的富马酸盐无定型或富马酸盐晶型a,在乙腈(简称acn)中(重量体积比为40g/l),室温下搅拌约3天后离心,除去上清液,室温(25℃)下干燥去除残留溶剂所得固体后即为富马酸盐晶型b。
[0610]
晶型b的xrpd结果如图76,衍射数据如下表21所示。
[0611]
tga结果(图77)显示样品加热至142.9℃时有1.5%的失重。
[0612]
dsc结果(图78)显示在123.7℃(峰值温度)有1个吸热峰。
[0613]1h-nmr结果(图79)显示样品中游离碱与富马酸的摩尔比为1:1.0,无残留溶剂。
[0614]
dvs结果(图80)显示样品在80%rh下增重4.28%,有引湿性。
[0615]
4. 富马酸盐晶型c富马酸盐晶型c可由上述方法制得的富马酸盐无定型或富马酸盐晶型a在乙酸乙酯中(重量体积比为40g/l)室温下搅拌约3天后,离心去除上清液,所得的固体干燥后即为富马酸盐晶型c。
[0616]
富马酸盐晶型c的xrpd结果如图81,衍射数据如下表22所示。
[0617]
tga结果(图82)显示样品加热至148.9℃时有0.78 %的失重。
[0618]
dsc结果(图83)显示在143.71℃(峰值温度)有1个吸热峰。
[0619]1h-nmr 结果(图84)显示样品中游离碱与富马酸的摩尔比为1:1.15,残留溶剂etoac与游离碱摩尔比为0.03 (对应失重0.4%)。
[0620]
dvs结果(图85-图86)显示样品在80%rh下增重2.04%,有引湿性,吸湿前后晶型未发生变化。
[0621]
5. 富马酸盐晶型d富马酸盐晶型d为水合物晶型,可由上述方法制得的富马酸盐无定型或富马酸盐晶型a在水(重量体积比为40g/l)中室温下搅拌约3天后,离心去除上清液,所得的固体干燥后即为富马酸盐晶型d。或者,将富马酸盐晶型a在水活度0.80以上(例如甲醇和水)的溶剂中在室温下搅拌2天以上也可得到。
[0622]
富马酸盐晶型d的xrpd结果如图87,衍射数据如下表23所示。
[0623]
tga结果(图88)显示样品加热至127℃时有2.31%的失重(对应一水合物失重为2.44%)。
[0624]
dsc结果(图89)显示在76.8,113.6和143.71℃(峰值温度)有3个吸热峰。
[0625]1h-nmr 结果(图90)显示样品中游离碱与富马酸的摩尔比为1:1.03。
[0626]
dvs结果(图91)显示样品在80%rh下吸湿增重3.42%,有引湿性。
[0627]
6. 富马酸盐晶型g富马酸盐晶型g由上述方法制得的富马酸盐无定型或富马酸盐晶型a在meoh-h2o=85/15(v/v,aw=0.355,aw为水活度)(重量体积比为40g/l)中,室温下,搅拌约3天后,离心去除上清液,所得的固体干燥后即为富马酸盐晶型g。
[0628]
富马酸盐晶型g的xrpd结果如图92,衍射数据如下表24所示。
[0629]
tga结果(图93)显示样品加热至150℃时有1.46%的失重。dsc结果(图94)显示在102.60℃和130.69℃(峰值温度)有2个吸热峰。
[0630]
7. 富马酸盐晶型j富马酸盐晶型j为乙腈溶剂合物,可由上述方法制得的富马酸盐无定型或上述制备方法制得的富马酸盐晶型a在acn中室温下搅拌约3天后离心直接进行湿品检测。
[0631]
晶型j的xrpd结果如图95,衍射数据如下表25所示。
[0632]
9. 富马酸盐多晶型筛选试验1. 悬浮液法取50 mg的富马酸盐无定型到2 ml的玻璃瓶中,加入搅拌子,然后分别添加200μl溶剂(如下表所示),所得混悬液在25℃或50℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表26。
[0633]
2. 挥发法
分别称取约30 mg的富马酸盐无定型至4 ml小瓶中,加入1ml溶剂(如下表27),使用0.22 μm的尼龙滤头过滤,用parafilm
®
封口膜封住小瓶,并在上面扎有针孔,放置在室温下缓慢挥发。收集所得固体并进行xrpd测试,其结果参见表27。
[0634]
3. 快速冷却法分别称取约30 mg的富马酸盐无定型至4ml小瓶中,加入1ml溶剂(如下表28),使用0.22μm的尼龙滤头过滤,放置在5℃冰箱内冷却。收集所得固体并进行xrpd测试,其结果参见表28。
[0635]
发明人在研发过程中发现在制备的多种盐的晶型中游离碱无定型或是富马酸盐的无定型在特定的不同溶剂中能够形成多种晶型,经检测,富马酸盐晶型a-d相对于晶型g和晶型j具有更低的引湿性。其中,对富马酸晶型a进行溶解度和固体稳定性的实验中发现,其同时还具备更高的溶解度和固体稳定性,是一种兼具低引湿性、高溶解性和固体稳定性
的晶型,在制备成药物制剂时相对于其他的晶型具有更高的生物利用度。
[0636]
实施例7 甲磺酸盐晶型的制备1. 甲磺酸盐晶型a甲磺酸盐晶型a由1g的游离碱无定型(由实施例1中方法制得的如式i化合物)与甲磺酸(酸碱投料摩尔比1:1)在25ml的ipac中室温下搅拌约3天后,离心去除上清液,得到的固体在室温下干燥过夜即得晶型a。
[0637]
xrpd结果如图96所示,衍射数据如下表29所示。
[0638]
tga结果(图97)显示样品加热至180℃时有3.3%的失重。
[0639]
dsc结果(图98)显示样品在175.3℃(起始温度)有1个吸热峰。
[0640]1h-nmr结果(图99)显示,样品中甲磺酸与游离碱摩尔比为0.9,残留溶剂ipac与游离碱摩尔比为0.03 (对应失重0.5%)。
[0641]
dvs结果(图100-图101)显示样品在80%rh下吸湿增重12.48%,吸湿前后晶型未发生变化。
[0642]
甲磺酸盐多晶型筛选试验1. 悬浮液法取50 mg的甲磺酸盐晶型a到2ml的玻璃瓶中,加入搅拌子,然后分别添加200μl溶剂(如下表所示),所得混悬液在40℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表30。
[0643]
实施例8 草酸盐晶型的制备1 草酸盐晶型a草酸盐晶型a可由游离碱无定型(由实施例1中方法制得的如式i化合物)与草酸(酸碱投料摩尔比1:1)分别在丙酮/正庚烷 (1:1,v/v)中室温下搅拌约3天后,离心去除上清液,干燥沉淀后所得的固体即为草酸盐晶型a。
[0644]
xrpd结果列于图102,衍射数据如下表31所示。
[0645]
tga结果(图103)显示样品加热至150℃时有6.7%的失重。
[0646]
dsc结果(图104)显示样品在85.5、172.5和190.8℃(峰值温度)有3个吸热峰。
[0647]1h-nmr结果(图105)显示样品中残留正庚烷与游离碱摩尔比为0.05 (对应失重0.7%),未检测到丙酮溶剂残留。
[0648]
uplc/ic 结果显示样品游离碱和草酸的摩尔比为1:1.2。
[0649]
2. 草酸盐晶型b草酸盐晶型b由游离碱无定型(由实施例1中方法制得的如式i化合物的游离碱无
定型)与草酸(酸碱投料摩尔比1:1)分别在ipac中室温下搅拌约3天后,离心去除上清液,干燥沉淀后所得的固体即为草酸盐晶型b。
[0650]
xrpd 结果列于图106,衍射数据如下表32所示。
[0651]
tga结果(图107)显示样品加热至150℃时有4.89%的失重。
[0652]
dsc结果(图108)显示样品在123.2和182.4℃(峰值温度)有2个吸热峰。
[0653]1h-nmr 结果(图109)显示样品中残留ipac与游离碱摩尔比为0.08(对应失重1.0%)。
[0654]
uplc/ic 结果显示样品游离碱和草酸的摩尔比为1:1.6。
[0655]
3. 草酸盐晶型c草酸盐晶型c由游离碱无定型(由实施例1中方法制得的如式i化合物)与草酸(酸碱投料摩尔比1:1)在thf 中室温下搅拌约3天后,离心去除上清液,干燥沉淀后所得的固体即为草酸盐晶型c。
[0656]
xrpd结果如图110所示,衍射数据如下表33所示。
[0657]
tga结果(图111)显示样品加热至150℃时有5.78%的失重。
[0658]
dsc结果(图112)在42.1、179.4和195.8℃(峰值温度)有3个吸热峰。
[0659]1h-nmr结果(图113)显示未检测到thf溶剂残留。
[0660]
uplc/ic结果显示样品酸碱摩尔比为1:1.7。
[0661]
实施例9 氢溴酸盐晶型的制备1 氢溴酸盐晶型a氢溴酸盐晶型a由游离碱无定型(由实施例1中方法制得的如式i化合物)与ipac混合后,加入氢溴酸(酸碱投料摩尔比为1:1)在室温下搅拌约3天后,离心去除上清液,干燥沉淀后所得的固体即为氢溴酸盐晶型a。
[0662]
xrpd结果列于图114,衍射数据如下表34所示。
[0663]
tga结果(图115)显示样品加热至150℃时有6.3%的失重。
[0664]
dsc结果(图116)显示样品在96.5和140.6℃(峰值温度)有2个吸热峰。
[0665]1h-nmr结果(图117)显示样品中残留ipac与api的摩尔比为0.01 (对应失重0.2%)。
[0666]
uplc/ic结果显示样品酸碱摩尔比为1.1。
[0667]
2. 氢溴酸盐晶型b氢溴酸盐晶型b由游离碱无定型(由实施例1中方法制得的如式i化合物)与ipac混合后,加入氢溴酸(酸碱投料摩尔比为2:1)在室温下搅拌约3天后,离心去除上清液,干燥沉淀后所得的固体即为氢溴酸盐晶型b。
[0668]
xrpd 结果列于图118,衍射数据如下表35所示。
[0669]
tga结果(图119)显示样品加热至150℃时有8.7%的失重。
[0670]
dsc结果(图120)显示样品在131.8、172.7和178.7℃(峰值温度)有3个吸热峰。
[0671]1h-nmr 结果(图121)显示样品中未检测出ipac溶剂残留。
[0672]
uplc/ic 结果显示样品游离碱和溴酸的摩尔比为1:1.8。
[0673]
3. 氢溴酸盐晶型c取1g的api(由实施例1中方法制得的如式i化合物的游离碱无定型)到40ml的玻璃瓶中,加入5ml的乙醇得到棕色溶液,150mg的氢溴酸溶液(2ml乙醇稀释)加入(式i化合物与氢溴酸的投料比为1:1.1),无沉淀析出,搅拌1小时后,加入~25ml的正庚烷,析出少量的固体,混悬液继续搅拌过夜。样品析出的固体很少,直接将样品旋干,得到溴酸盐晶型c。
[0674]
xrpd结果如图122,衍射数据如下表36所示。
[0675]
tga结果(图123)显示样品加热至149℃时有3.26% 的失重。
[0676]
dsc结果(图124)显示样品在50.98和117.20℃(峰值温度)有2个吸热峰。
[0677]
dvs结果(图125)显示样品在80%rh下吸湿增重6.97%。
[0678]
4. 氢溴酸盐多晶型筛选悬浮液法:取50mg采用上述方法制得的氢溴酸盐晶型a加入到2ml的玻璃瓶中,加入搅拌子,然后分别添加200μl溶剂(如下表所示),所得混悬液在25℃下搅拌4天后,快速离心,取残余固体于真空干燥箱中(-0.1mpa,25℃)干燥。其结果参见表37。
[0679]
发明人在研发过程中发现游离碱无定型在特定的不同溶剂或是不同的酸碱投料比下与氢溴酸的混合液能够形成不同的氢溴酸盐晶型,其中,三种晶型中氢溴酸盐晶型c的引湿性最低,且热稳定性好,同时,其在不同生物溶媒中均具有较佳地溶解度,由此可见,氢溴酸盐晶型c在制备成药物制剂时,相对于无定型以及其他氢溴酸盐晶型具有更高的生物利用度。
[0680]
溶解度试验对游离碱晶型a和6种盐型在水和3种生物溶媒中的动态溶解度进行了评估。
[0681]
称取约9~20mg物料(游离碱或盐晶型)于5ml小瓶中,加入3~4ml溶剂,在37℃下旋转混合2和24小时(500rpm),取样约0.8ml离心过滤,液体测试浓度。溶解度结果如表38所示。
[0682]
结果显示在水和3种生物溶媒中,6种盐型的溶解度均大于游离碱晶型a。
[0683]
固体稳定性试验将游离碱无定型和3种盐型分别在25℃/60%rh、40℃/75%rh和50℃/75%rh条件下放置1周和2周后,分别取样进行检测(hplc,xrpd)。
[0684]
稳定性数据结果(表39)显示:结果显示游离碱无定型在40℃/75%rh和50℃/75%rh条件下潮解。
[0685]
甲磺酸盐晶型a在40℃/75%rh条件下放置1周后转为新晶型,其他样品(游离碱无定型、硫酸盐晶型a、富马酸盐晶型a)在放置后晶型不变。
[0686]
游离碱无定型在25℃/60%rh下放置2 周后纯度无明显变化,40℃/75%rh和50℃/
75%rh条件下 放置1周后纯度下降。
[0687]
硫酸盐晶型a在25
ꢀº
c/60%rh和40℃/75%rh条件下放置2 周后纯度无明显变化,在50℃/75%rh条件下放置1周后纯度下降。
[0688]
富马酸盐晶型a在25℃/60%rh条件下放置2周后纯度无明显变化,在40℃/75%rh条件下放置2周和在50℃/75%rh条件下放置1周后纯度下降。
[0689]
甲磺酸盐晶型a在25℃/60%rh条件下放置2周后纯度无明显变化,在40℃/75%rh和50℃/75%rh条件下放置1周后纯度下降。
[0690]
根据上述实施例中所示的多种游离碱和盐的晶型以及无定型的检测结果可知,即使是制备出晶型结构,其也可能存在稳定性差、或者引湿性低等难以制备出生物利用度较高的药物制剂。发明人通过创造性的劳动进行的上述实验中发现,游离碱晶型a和富马酸盐晶型a的引湿性相对于其他晶型较低,仅为2%以下,引湿性相对较低的晶型还有盐酸盐晶型a、硫酸盐晶型c、马来酸盐晶型c、富马酸盐晶型a/b/c、以及氢溴酸盐晶型c,这些盐的晶型的引湿性均在8%以下,低于游离碱无定型。同时,根据溶解度实验可知,虽然多种晶型(硫酸
盐晶型a、富马酸盐晶型a、氢溴酸盐晶型a、盐酸盐晶型a和马来酸盐晶型a)的引湿性高于游离碱晶型a,但是在多种生物溶媒中具有较高的溶解度,并且固体稳定性也较高,在制备成药物制剂时仍然具备较高的优势。值得一提的是,富马酸盐晶型a不仅具有较低的引湿性还具有较佳地溶解度而且固体稳定性也较高,有望通过得到对癌症具有更强治疗效果的晶型化合物。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
