一种l-α-甘油磷酰胆碱中微量位置异构体杂质的检测方法
技术领域
1.本发明属于l-α-甘油磷酰胆碱分析检测领域,具体涉及一种定量核磁共振磷谱法检测l-α-甘油磷酰胆碱(简称l-α-gpc)中微量位置异构体杂质β-甘油磷酰胆碱(简称β-gpc)的方法。
背景技术:
2.有关物质是药物质量控制的关键指标,主要包括了反应起始物、中间体、副产物及降解物质等。为了保证药物在临床应用中的质量安全性,各国监管机构及人用药品注册技术要求国际协调会议(international council for harmonization,ich)均制定了相关的指导原则,通常规定有关物质的控制限度为0.1%。
3.l-α-甘油磷酰胆碱(l-α-glycerylphosphorylcholine,l-α-gpc)的结构式如下:
[0004][0005]
l-α-gpc作为人的机体内源性的水溶性小分子物质,能够有效预防和治疗阿尔兹海默氏症与多发性脑梗死性痴呆等神经性疾病,目前已在意大利、韩国、德国等多个国家上市。
[0006]
l-α-gpc的有关物质主要包括了起始物料磷酰胆碱(pc),降解产物甘油磷酸酯(gp)以及副产物l-α-甘油磷酰乙醇胺(l-α-gpe)和β-甘油磷酰胆碱(β-gpc)。由于l-α-gpc及其有关物质具有极性大,难挥发且无紫外吸收等特点,文献中主要采用了高效液相色谱法,并联用示差折光检测器或者蒸发光散射检测器对其有关物质进行检测。其中值得关注的是,β-gpc作为l-α-gpc位置异构体,与l-α-gpc并没有实现很好的色谱分离,给微量β-gpc的准确定量带来挑战。因此需要建立一种可以准确测定l-α-gpc中微量位置异构体杂质的检测方法。
[0007]
β-gpc结构式如下:
[0008]
技术实现要素:
[0009]
发明人在现有技术的基础上试验了许多种试剂、溶剂和实验参数,发现只有在特定的试剂、溶剂和参数的情况下才能准确定量并完全分离β-gpc与l-α-gpc的谱峰,并在此基础上完成了本发明。因此,本发明的目的是提供一种定量核磁共振磷谱法,可以使β-gpc与l-α-gpc的谱峰完全分离,能够实现l-α-gpc中微量位置异构杂质β-gpc的准确定量,由此可以确保gpc产品的质量。
[0010]
为实现以上目的,本发明采用的技术方案为:
[0011]
一种使用核磁共振波谱仪通过定量核磁共振磷谱检测l-α-gpc中位置异构体杂质β-gpc的方法,其特征在于,该方法使用氧化氘作为溶剂,且使用以磷酸三甲酯作为内标物的内标法,或以β-gpc为对照品的外标法。
[0012]
根据本发明的一个实施方式,其中
[0013]
所述内标法包括:
[0014]
步骤1:称取内标物磷酸三甲酯溶于氧化氘中摇匀,得内标储备液备用;
[0015]
步骤2:称取样品溶于氧化氘中,加入内标储备液,再用氧化氘定容,得样品溶液备用;
[0016]
步骤3:取步骤2中的样品溶液采集核磁信号,获得的β-gpc和磷酸三甲酯峰面积采用以下内标法公式(1)计算β-gpc的含量:
[0017][0018]
式中:i
β
为β-gpc的含量;as、ar分别为β-gpc和内标物的积分峰面积;cs、cr分别为样品溶液中样品配制浓度和内标物的配制浓度,单位为g
·
l-1
;ms、mr分别为β-gpc和内标物的摩尔质量;pr为内标物的纯度;
[0019]
所述外标法包括:
[0020]
步骤1’:称取β-gpc溶于氧化氘中摇匀,得外标溶液备用;
[0021]
步骤2’:称取样品溶于氧化氘中摇匀,得样品备用;
[0022]
步骤3’:取步骤1’中的外标溶液和步骤2’中的样品溶液分别采集核磁信号,获得的β-gpc峰面积采用外标法公式(2)计算β-gpc的含量:
[0023][0024]
式中:i
β
为β-gpc的含量;as、ar分别为样品溶液和β-gpc对照品溶液中β-gpc的积分峰面积;cs、cr分别为样品溶液的配制浓度和β-gpc对照品溶液的配制浓度,单位为g
·
l-1
;pr为β-gpc对照品的纯度。
[0025]
根据本发明的一个实施方式,其中,所述核磁共振波谱仪使用的脉冲序列为zgpg去耦模式;
[0026]
根据本发明的一个实施方式,其中,所述核磁共振波谱仪的仪器探头为pabbo或cppbbo。
[0027]
根据本发明的一个实施方式,其中,所述核磁共振波谱仪的弛豫时间为1~60s;
[0028]
根据本发明的一个实施方式,其中,所述核磁共振波谱仪的扫描次数为1~200次,例如为40次。
[0029]
根据本发明的一个实施方式,其中,所述核磁共振波谱仪的弛豫时间为40s。
[0030]
根据本发明的一个实施方式,其中,以化学位移为-0.85ppm的特征峰为β-gpc的定量峰。
[0031]
有益效果
[0032]
经专属性、线性与范围、定量限、检测限、重复性和准确度试验对本发明的方法进行了方法学验证,证明本发明提供的一种l-α-gpc中微量位置异构杂质β-gpc的检测方法具有下述优点:
[0033]
(1)在采用特定的条件参数时,例如,采用5mm cppbbo探头测定;温度:298k;谱宽:187.5khz;脉冲序列:zgpg;脉冲宽度:12μs;采样时间:1s;弛豫时间:40s;扫描次数:40次的参数下采集上述混合溶液信号时,l-α-gpc及其含磷杂质(l-α-gpe、pc及gp)的谱峰均与位置异构体杂质β-gpc及内标磷酸三甲酯谱峰完全分离(图1),满足定量测定要求,表明本方法具有良好的专属性。
[0034]
(2)β-gpc在一定浓度范围内,呈现良好的线性关系。
[0035]
(3)本方法具有良好的重复性和准确度。
[0036]
(4)本方法的检测限为0.01g
·
l-1
,表明灵敏度较高,能够满足微量杂质的定量要求。
[0037]
(5)无复杂的样品预处理。
[0038]
本发明首次提供了一种核磁共振磷谱法测定l-α-gpc中微量位置异构体含量的方法,为提升l-α-gpc中位置异构体的含量控制提供重要参考依据。
附图说明
[0039]
图1是采用本发明的方法检测l-α-甘油磷酰胆碱及其有关物质与内标物磷酸三甲酯的核磁共振磷谱图。图中各峰代表的是:1.磷酸三甲酯,2~3.甘油磷酸酯,4.l-α-甘油磷酰乙醇胺,5.l-α-甘油磷酰胆碱,6.磷酰胆碱,7.β-甘油磷酰胆碱,*.未知杂质。
[0040]
图2是分别采用氘代甲醇和氧化氘为溶剂测得的样品溶液对比图。图中各峰代表的是:1.磷酸三甲酯,2.l-α-甘油磷酰胆碱,3.β-甘油磷酰胆碱。
具体实施方式
[0041]
下面详细描述本发明的实施例,需要说明的是下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。另外,如果没有明确说明,在实施例中采用的试剂均为市场上可获得。
[0042]
实施例1:
[0043]
本实施例提供了采用本发明对l-α-gpc中位置异构体的含量进行检测的方法,使用内标法进行定量计算。
[0044]
仪器与参数:
[0045]
bruker biospin gmbh 500mhz核磁共振波谱仪;5mm cppbbo探头;温度:298k;谱宽:187.5khz;脉冲序列:zgpg;脉冲宽度:12μs;采样时间:1s;弛豫时间:40s;扫描次数:40次。
[0046]
实验步骤
[0047]
步骤1:内标储备液:称取内标物磷酸三甲酯0.015g,用氧化氘溶解并稀释至25ml,摇匀,备用。
[0048]
步骤2:样品溶液:称取样品0.5g,加少量氧化氘溶解,再精密加入0.50ml内标储备液,用氧化氘稀释至5ml,摇匀,即得。
[0049]
取样品溶液0.5ml,置5mm核磁管中,在上述仪器参数下采集信号,进行相位调整和基线校正后进行积分,获得的β-gpc和磷酸三甲酯峰面积采用内标法公式(1)计算β-gpc的含量。
[0050][0051]
式中:i
β
为β-gpc的含量;as、ar:分别为β-gpc和内标物的积分峰面积;cs、cr:分别为样品溶液中样品配制浓度和内标物的配制浓度(g
·
l-1
);ms、mr:分别为β-gpc和内标物的摩尔质量;pr为内标物的纯度。
[0052]
试验结果显示,内标物磷酸三甲酯、位置异构体杂质β-gpc与l-α-gpc三者谱峰完全分离,检出的β-gpc含量为0.06%,小于0.1%,表明本发明方法可以实现对l-α-gpc中微量位置异构体杂质β-gpc量的准确测定。
[0053]
实施例2
[0054]
除了使用pabbo探头代替cppbbo探头,扫描次数80次代替40次以外,以与实施例1相同的方式测量样品溶液的微量位置异构体杂质β-gpc量。结果β-gpc的量为0.06%,与实施例1的结果一致。
[0055]
实施例3
[0056]
本实施例使用实施例1中的样品,使用外标法对l-α-gpc中的β-gpc进行定量计算。
[0057]
实验步骤
[0058]
β-gpc对照品溶液:称取β-gpc对照品0.01g,用氧化氘溶解并稀释至10ml,摇匀,得β-gpc对照品储备液;量取β-gpc对照品储备液0.50ml,用氧化氘稀释至5ml,摇匀,即得。
[0059]
样品溶液:称取样品0.5g,加氧化氘溶解并稀释至5ml,摇匀,即得。
[0060]
采用实施例1的仪器参数对β-gpc对照品溶液和样品溶液采集信号,进行相位调整和基线校正后进行积分,获得的β-gpc峰面积采用外标法公式(2)计算β-gpc的含量。
[0061][0062]
式中:i
β
为β-gpc的含量;as、ar:分别为样品溶液和β-gpc对照品溶液中β-gpc的积分峰面积;cs、cr:分别为样品溶液的配制浓度和β-gpc对照品溶液的配制浓度(g
·
l-1
);pr为β-gpc对照品的纯度。
[0063]
试验结果显示,检出的β-gpc含量为0.06%,与实施例1的结果一致。表明本发明方法可采用内标法或外标法对l-α-gpc中微量位置异构体杂质β-gpc量的准确测定。
[0064]
实施例4
[0065]
本实施例通过添加工艺过程中可能存在的其他含磷杂质考察本发明方法的专属性,同时采用β-gpc对照品考察本发明方法的线性范围和准确度。
[0066]
溶液配制:
[0067]
内标储备液:称取内标物磷酸三甲酯0.015g,用氧化氘溶解并稀释至25ml,摇匀,得内标储备液备用;
[0068]
样品溶液:称取样品0.5g,加少量氧化氘溶解,再精密加入0.5ml内标储备液,用氧化氘稀释至5ml,摇匀,得样品溶液备用;
[0069]
β-gpc对照品储备液:称取β-gpc对照品0.01g,用氧化氘溶解并稀释至10ml,摇匀,得β-gpc对照品储备液备用;
[0070]
混合溶液:分别称取样品、各个含磷杂质(β-gpc、l-α-gpe、pc、gp)和内标物磷酸三甲酯适量,用氧化氘溶解并稀释制成样品100g
·
l-1
,含磷杂质和磷酸三甲酯各0.1g
·
l-1
的
混合溶液;
[0071]
线性溶液:分别量取β-gpc对照品储备液0.16ml、0.25ml、0.40ml、0.50ml、0.60ml和1.0ml,同时各加入0.5ml内标储备液,用氧化氘稀释至5ml,摇匀,得线性溶液;
[0072]
回收率溶液:取样品9份,分为3组,每组分别加入内标储备液和3个不同浓度水平的β-gpc对照品储备液,用氧化氘稀释制成含样品100g
·
l-1
,依次含已知浓度的β-gpc为0.05g
·
l-1
、0.08g
·
l-1
、0.10g
·
l-1
,以及含磷酸三甲酯0.06g
·
l-1
的混合溶液。
[0073]
采用实施例1的仪器参数对上述的混合溶液、线性溶液和准确度溶液进行信号采集,结果发现l-α-gpc及其含磷杂质(l-α-gpe、pc及gp)的谱峰均与β-gpc、磷酸三甲酯谱峰完全分离,满足定量测定要求,表明本法具有良好的专属性。
[0074]
以β-gpc与磷酸三甲酯的质量浓度比值为横坐标(x),峰面积比值为纵坐标(y)进行线性回归,获得线性方程为y=0.5439x-0.027(n=6,r=0.9997),结果表明β-gpc在0.03~0.20g
·
l-1
的浓度范围内具有良好的线性关系。
[0075]
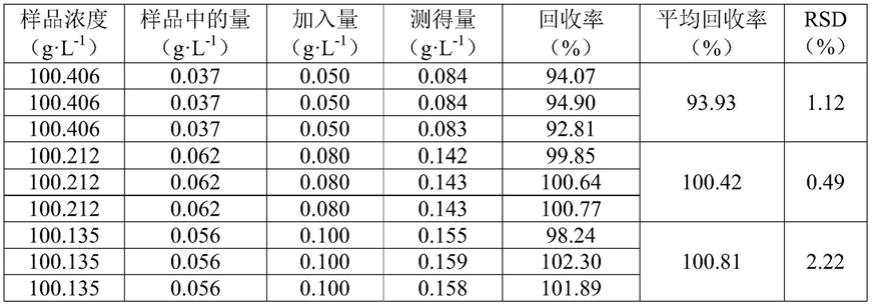
回收率考察结果如表1所示,其中样品中的量和测得量均采用实施例1中的公式(1)获得。本方法的回收率均在92%~103%之内,表明本发明的准确度良好,能够准确测定l-α-gpc中β-gpc的含量。
[0076]
表1回收率测定结果
[0077][0078]
对比实施例1
[0079]
除了使用氘代甲醇(meod)代替氧化氘(d2o)以外,以与实施例1相同的方式测量样品溶液的微量位置异构体杂质β-gpc量。结果显示,使用氘代甲醇时l-α-gpc(δ0.02ppm)与β-gpc(δ-0.44ppm)不能完全分离(图2),从而无法对β-gpc进行准确定量。
[0080]
对比实施例2
[0081]
除了使用磷酸吡哆醛与磷酸三乙酯代替磷酸三甲酯作为内标物以外,以与实施例1相同的方式测量样品溶液的微量位置异构体杂质β-gpc量。结果发现磷酸吡哆醛(δ0.03ppm)和磷酸三乙酯(δ-0.43ppm)均与l-α-gpc(δ-0.14ppm)未能完全分离,无法作为内标物对β-gpc进行定量检测。
[0082]
根据实施例1和对比实施例1的结果可以看出,在氘代试剂条件不满足的情况下,会发生β-gpc位移向l-α-gpc靠近的情况,导致两谱峰不能完全分离,从而无法对β-gpc准确定量。因此只有同时满足特定的试剂、溶剂和参数的情况下才能准确定量并完全分离β-gpc与l-α-gpc的谱峰。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。