1.本发明涉及一种聚乙醇酸复合材料及其制备方法和改性剂的用途。
背景技术:
2.聚乙醇酸(polyglycolicacid,pga)因其良好生物降解性、生物相容性及水汽阻隔性优异而备受关注。但是,聚乙醇酸的结晶度过高,加工温度过高,韧性差,从而限制了pga的应用。
3.科技工作者尝试对聚乙醇酸进行改性。例如,cn111454552a公开了聚乙醇酸组合物,包括60~95重量份聚乙醇酸和1.5~18重量份聚乙醇酸改性剂,其中,聚乙醇酸改性剂包括组分a和组分b,组分a包括稀土助剂1~15重量份;组分b包括:扩链剂0.1~3重量份、成核剂0.05~1重量份、引发剂0.1~3重量份、抗氧剂0.05~1重量份。又如,cn113088055a公开了一种聚乙醇酸基复合材料,包括聚乙醇酸50~95重量份,可降解聚酯5~50重量份,功能填料0.5~30重量份,相容剂0.01~10重量份,抗氧剂0~3重量份,润滑剂0~1重量份,扩链剂0~1重量份。
4.超支化聚合物是一种以ab2型单体分子为生长点,通过逐步控制重复化学反应的方式得到一系列分子量不断增长且结构相似的化合物。超支化聚合物具有高度支化的三维交联网状结构和大量的活性端基,具有良好的溶解性、低熔体粘度。超支化聚合物因其独特性质日益受到人们关注。cn110951166a公开了超支化聚合物改性聚丙烯以提高其刚性。cn113564748a公开了超支化聚合物与聚碳酸酯纤维膜复合得到力学性能提升的聚碳酸酯纤维复合膜。cn107824057a公开了一种超支化聚合物改性抗污聚合物薄膜,其抗污染能力、抗菌性能得到提升。
5.目前为止,仍未有关于超支化聚合物改善聚乙醇酸韧性的报道。
技术实现要素:
6.有鉴于此,本发明的一个目的在于提供一种聚乙醇酸复合材料。与纯聚乙醇酸树脂相比,本发明的聚乙醇酸复合材料的韧性得到明显改善,尤其是断裂伸长率明显提高。本发明的另一个目的在于提供一种聚乙醇酸复合材料的制备方法。本发明的再一个目的在于提供一种超支化聚合物改性剂的用途。本发明采用如下技术方案实现上述目的。
7.一方面,本发明提供一种聚乙醇酸复合材料,所述聚乙醇酸复合材料包括:
8.聚乙醇酸80~95重量份,和
9.超支化聚合物改性剂0.8~10重量份;
10.其中,所述超支化聚合物改性剂由式(a)所示的化合物与二元胺反应而得,所述二元胺选自c3-c38脂肪族二元胺、c6-c30脂环族二元胺或c6-c20芳香族二元胺;
c6烷氧基,且r1和r2至少有一个为氢。
24.根据本发明所述的聚乙醇酸复合材料,优选地:
25.式(a)中,x为氯;
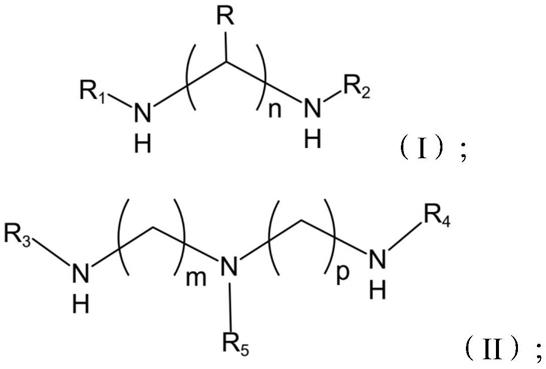
26.式(i)中,3≤n≤5;r选自氢、c1-c3烷基或c1-c3烷氧基,且r1和r2均为氢;
27.式(ii)中,m、p相同,且3≤m≤5,3≤p≤5;r3、r4均为氢;r5选自c1-c3烷基或c1-c3烷氧基;
28.式(iii)中,r6、r7、r8和r9独立地选自氢、c1-c3烷基或c1-c3烷氧基;
29.式(iv)中,r1和r2均为氢。
30.根据本发明所述的聚乙醇酸复合材料,优选地,所述聚乙醇酸复合材料包括:
31.聚乙醇酸90~95重量份,和
32.超支化聚合物改性剂1~7重量份。
33.根据本发明所述的聚乙醇酸复合材料,优选地,还包括:
34.环氧大豆油1~10重量份。
35.根据本发明所述的聚乙醇酸复合材料,优选地,还包括:
36.柠檬酸三丁酯1.5~7重量份。
37.另一方面,本发明提供根据如上所述的聚乙醇酸复合材料的制备方法,包括以下步骤:
38.1)将包括聚乙醇酸和超支化聚合物改性剂的原料混合均匀,得到混合物;将混合物用双螺杆挤出机进行熔融剪切混炼,得到混炼产物;
39.2)将混炼产物采用注塑机注塑成型,得到聚乙醇酸复合材料;
40.其中,双螺杆挤出机包括上腔板、双螺杆和下腔板,且所述双螺杆位于上腔板和下腔板之间,所述上腔板位于所述双螺杆的上方;所述上腔板的温度为220~230℃,所述下腔板的温度为225~235℃;
41.其中,注塑机包括料筒和空腔,所述空腔位于所述料筒的下方;注塑拉伸样条模具位于所述空腔内;料筒内温度为200~220℃,注塑拉伸样条模具内的温度为80~160℃。
42.根据本发明所述的制备方法,优选地,双螺杆挤出机的螺杆转速为29~50rpm。
43.根据本发明所述的制备方法,优选地,所述超支化聚合物改性剂通过如下方法获得:
44.1-1)将所述二元胺、催化剂和水混合,形成第一溶液;所述催化剂选自碳酸钠、碳酸钾、三乙胺、三乙醇胺和吡啶中的至少一种;
45.1-2)将第一溶液滴加至含所述式(a)所示的化合物的第二溶液中反应,得到反应产物;其中,第二溶液所用溶剂选自丙酮、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、四氢呋喃和1,4-二氧六环中的一种或者多种;
46.1-3)将反应产物在6000~10000rpm的转速下离心5~15min得到固体产物,将固体产物依次用水和丙酮淋洗,然后真空干燥,得到超支化聚合物改性剂。
47.再一方面,本发明还提供一种超支化聚合物改性剂在提高聚乙醇酸复合材料的韧性中的用途,所述超支化聚合物改性剂由式(a)所示的化合物与二元胺反应而得;所述二元胺选自c3-c38脂肪族二元胺、c6-c30脂环族二元胺或c6-c20芳香族二元胺;
[0048][0049]
式(a)中,x为氯、溴或碘。
[0050]
与纯聚乙醇酸树脂相比,本发明的聚乙醇酸复合材料的韧性得到明显改善,尤其是断裂伸长率得到明显提高。根据本发明优选的技术方案,通过控制复合材料中超支化聚合物改性剂的含量,可以进一步提高聚乙醇酸复合材料的韧性。
具体实施方式
[0051]
下面结合具体实施例对本发明作进一步的说明,但本发明的保护范围并不限于此。
[0052]
《聚乙醇酸复合材料》
[0053]
本发明的聚乙醇酸复合材料包括聚乙醇酸和超支化聚合物改性剂。优选地,聚乙醇酸复合材料包括聚乙醇酸、超支化聚合物改性剂以及环氧大豆油和/或柠檬酸三丁酯。
[0054]
根据本发明的一个实施方式,聚乙醇酸复合材料包括聚乙醇酸、超支化聚合物改性剂和环氧大豆油。根据本发明的另一个实施方式,聚乙醇酸复合材料包括聚乙醇酸、超支化聚合物改性剂和柠檬酸三丁酯。根据本发明的再一个实施方式,聚乙醇酸复合材料包括聚乙醇酸、超支化聚合物改性剂、环氧大豆油和柠檬酸三丁酯。环氧大豆油和柠檬酸三丁酯的重量比为1:1~2,优选为1:1.2~1.7,更优选为1: 1.3~1.5。
[0055]
根据本发明的一个实施方式,聚乙醇酸复合材料由聚乙醇酸、超支化聚合物改性剂和环氧大豆油组成。根据本发明的另一个实施方式,聚乙醇酸复合材料由聚乙醇酸、超支化聚合物改性剂和柠檬酸三丁酯组成。根据本发明的再一个实施方式,聚乙醇酸复合材料由聚乙醇酸、超支化聚合物改性剂、环氧大豆油和柠檬酸三丁酯组成。环氧大豆油和柠檬酸三丁酯的重量比为1:1~2,优选为1:1.2~1.7,更优选为1:1.3~1.5。
[0056]
聚乙醇酸的用量可以为80~95重量份,优选为85~95重量份,更优选为90~95重量份。聚乙醇酸的数均分子量可以为10000~ 60000,优选为10000~50000,更优选为10000~40000,再优选为 20000~40000。
[0057]
超支化聚合物改性剂的用量可以为0.8~10重量份,优选为1~7 重量份,更优选为2.5~7重量份。这样有利于提高聚乙醇酸复合材料的韧性,尤其是断裂伸长率。
[0058]
在本发明中,超支化聚合物改性剂由式(a)所示的化合物与二元胺反应而得到。式(a)所示的化合物的结构式如下:
[0059][0060]
式(a)中,x为氯、溴或碘。优选地,x为氯或溴。更优选地,x为氯。式(a)所示的化合
物的实例包括但不限于三聚氯氰或三聚溴氰。
[0061]
本发明所述的二元胺选自c3-c38脂肪族二元胺、c6-c30脂环族二元胺或c6-c20芳香族二元胺。优选地,所述的二元胺选自c3-c38 脂肪族二元胺或c6-c30脂环族二元胺。更优选地,所述的二元胺为 c3-c38脂肪族二元胺。c3-c38、c6-c30和c6-c20分别表示碳原子数。
[0062]
在本发明中,脂肪族二元胺的碳原子数可以为c3-c38,优选为 c3-c30,更优选为c3-c20,再优选为c3-c15。
[0063]
在本发明中,脂肪族二元胺可以为式(i)所示的化合物或式(ii)所示的化合物。
[0064]
式(i)所示的化合物结构式如下:
[0065][0066]
式(i)中,n为自然数,且3≤n≤8;优选地,3≤n≤5;更优选地,n为3或4;再优选地,n为3。r、r1、r2独立地选自氢、c1
‑ꢀ
c6烷基或c1-c6烷氧基,且r1和r2至少有一个为氢。
[0067]
式(i)中,c1-c6烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基和正己基。 c1-c6烷氧基的实例包括但不限于甲氧基、乙氧基、丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、正己氧基。
[0068]
r优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r为氢。
[0069]
r1优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r1为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r1为氢。
[0070]
r2优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r2为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r2为氢。
[0071]
式(ii)所示的化合物结构式如下:
[0072][0073]
式(ii)中,m、p均为自然数,m、p相同或不同,且2≤m≤6,2≤p≤6;优选地,m、p相同,且3≤m≤5,3≤p≤5;更优选地,m、p 相同,且m、p均为3或4。r3、r4独立地选自氢、c1-c6烷基或 c1-c6烷氧基,且r3和r4至少有一个为氢;r5选自c1-c6烷基或 c1-c6烷氧基。
[0074]
式(ii)中,c1-c6烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基和正己基。 c1-c6烷氧基的实例包括但不限于甲氧基、乙氧基、丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、正己氧基。
[0075]
r3优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r3为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r3为氢。
[0076]
r4优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r4为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r4为氢。
[0077]
r5优选选自c1-c3烷基或c1-c3烷氧基。更优选地,r5为甲基、乙基或甲氧基。根据本发明的一个具体实施方式,r5为甲基。
[0078]
本发明的脂环族二元胺的碳原子数可以为c6-c30,优选为c6
‑ꢀ
c20,更优选为c6-c12。
[0079]
脂环族二元胺可以为式(iii)所示的化合物。式(iii)所示的化合物结构式如下:
[0080][0081]
式(iii)中,r6、r7、r8和r9独立地选自氢、c1-c6烷基或c1-c6烷氧基。式(iii)中,c1-c6烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基和正己基。c1-c6烷氧基的实例包括但不限于甲氧基、乙氧基、丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、正己氧基。
[0082]
r6优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r6为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r6为氢。
[0083]
r7优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r7为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r7为氢。
[0084]
r8优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r8为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r8为氢。
[0085]
r9优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r9为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r9为氢。
[0086]
根据本发明优选的一个具体实施方式,r6、r7、r8和r9均为氢。
[0087]
本发明的式(iii)所示的化合物可以为顺反混合物的二元胺,也可以为反式的二元胺。
[0088]
本发明所述的芳香族二元胺的碳原子数可以为c6-c20,优选为 c6-c16,更优选为c6-c10。
[0089]
芳香族二元胺可以为式(iv)所示的化合物。式(iv)所示的化合物结构式如下:
[0090][0091]
式(iv)中,两胺基为间位关系或对位关系;r1和r2独立地选自氢、c1-c6烷基或c1-c6烷氧基,且r1和r2至少有一个为氢。
[0092]
式(iv)中,c1-c6烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异
丁基、仲丁基、叔丁基、正戊基和正己基。 c1-c6烷氧基的实例包括但不限于甲氧基、乙氧基、丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、正己氧基。
[0093]
r1和r2独立地选自氢、c1-c6烷基或c1-c6烷氧基,且r1和r2至少有一个为氢。
[0094]
r1优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r1为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r1为氢。
[0095]
r2优选选自氢、c1-c3烷基或c1-c3烷氧基。更优选地,r2为氢、甲基或甲氧基。根据本发明的一个具体实施方式,r2为氢。
[0096]
根据本发明的一个具体实施方式,式(iv)所示的化合物为对苯二胺。
[0097]
本发明发现,上述式(i)所示的化合物、式(ii)所示的化合物、式(iii)所示的化合物或式(iv)所示的化合物与式(a)反应所得的超支化聚合物可以用于改善聚乙醇酸的韧性,尤其是提高聚乙醇酸复合材料的断裂伸长率。
[0098]
在本发明中,所述的二元胺的具体实例包括但不限于1,3-二氨基丙烷,2-甲基-1,3-二氨基丙烷,1,2,3-三甲基-1,3-二氨基丙烷,1,4-二氨基丁烷,1,5-二氨基戊烷,n,n-双(3-氨乙基)甲胺,n,n-双(3-氨丙基)甲胺,n,n-双(3-氨丙基)乙胺,n,n-双(3-氨丙基)丙胺,n,n-双(3
‑ꢀ
氨丁基)甲胺,n,n-双(3-氨丁基)乙胺,n,n-双(3-氨丁基)并胺,反式
‑ꢀ
1,4-环己二胺,1,4-环己二胺顺反混合物,2,6-二甲基-1,4-环己二胺,间苯二胺,对苯二胺。
[0099]
根据本发明优选的一个具体实施方式,所述的二元胺的实例为 1,3-二氨基丙烷,1,4-二氨基丁烷,n,n-双(3-氨丙基)甲胺,反式-1,4
‑ꢀ
环己二胺,1,4-环己二胺顺反混合物,对苯二胺。
[0100]
环氧大豆油的用量为1~10重量份,优选为1.5~5重量份,更优选为2~3重量份。这样可以增加聚乙醇酸和超支化聚合物改性剂的相容性,从而提高复合材料的韧性。
[0101]
柠檬酸三丁酯的用量为1.5~7重量份,优选为1.5~5重量份,更优选为2.5~4.5重量份。这样可以增加聚乙醇酸和超支化聚合物改性剂的相容性,从而提高复合材料的韧性。
[0102]
根据本发明的一个实施方式,聚乙醇酸复合材料包括80~95重量份聚乙醇酸,1~10重量份环氧大豆油和0.8~10重量份超支化聚合物改性剂。优选地,聚乙醇酸复合材料包括90~95重量份聚乙醇酸,1.5~5重量份环氧大豆油和2~7重量份超支化聚合物改性剂。
[0103]
根据本发明的另一个实施方式,聚乙醇酸复合材料包括85~95 重量份聚乙醇酸,1.5~7重量份柠檬酸三丁酯和1~8重量份超支化聚合物改性剂。优选地,聚乙醇酸复合材料包括90~95重量份聚乙醇酸,1.5~5重量份柠檬酸三丁酯和2~7重量份超支化聚合物改性剂。
[0104]
根据本发明的再一个实施方式,聚乙醇酸复合材料包括90~95 重量份聚乙醇酸,1.5~5重量份由重量比为1:1~2的环氧大豆油和柠檬酸三丁酯组成的混合物以及2~7重量份超支化聚合物改性剂。
[0105]
本发明的复合材料还可以包括抗氧剂、消泡剂、催干剂、稳定剂中的一种或多种。抗氧剂、消泡剂、催干剂、稳定剂可以为本领域已知的那些,在此不做赘述。
[0106]
《聚乙醇酸复合材料的制备方法》
[0107]
本发明的聚乙醇酸复合材料的制备方法包括以下步骤:1)将聚乙醇酸、助剂及超支化聚合物改性剂混合均匀,得到混合物;将混合物用双螺杆挤出机进行熔融剪切混炼,得
到混炼产物;2)将混炼产物注塑成型,得到聚乙醇酸复合材料。此外,本发明的制备方法还可以包括超支化聚合物改性剂的制备步骤。下面进行详细描述。
[0108]
超支化聚合物改性剂的制备步骤
[0109]
首先形成第一溶液,然后将第一溶液滴加至含三聚卤氰(详见前述式(a)所示的化合物)的第二溶液中,再经过后处理得到超支化聚合物改性剂。
[0110]
将如上所述的二元胺、催化剂和水混合形成第一溶液。二元胺的具体实例如前所述,这里不再赘述。
[0111]
本发明的第一溶液的溶剂可以为水,优选为去离子水。
[0112]
本发明的催化剂可以选自碳酸钠、碳酸钾、三乙胺、三乙醇胺和吡啶中的至少一种。优选地,催化剂选自碳酸钠、三乙胺和吡啶中的至少一种。更优选地,催化剂选自碳酸钠、三乙胺和吡啶中的一种。这样所得的超支化聚合物改性剂有利于提高聚乙醇酸复合材料的韧性。
[0113]
本发明的催化剂与所述的二元胺的摩尔比可以为0.25~0.35:1,优选为0.28~0.35:1,更优选为0.29~0.32:1。第一溶液中,二元胺的重量百分数为35~55wt%,优选为40~50wt%,更优选为42~ 49wt%。
[0114]
根据本发明的一个具体实施方式,将所述的二元胺、催化剂和水混合,在搅拌条件下加热至50~80℃,继续搅拌10~20min。优选地,加热至50~65℃,继续搅拌15~20min。这样有利于形成均匀稳定的第一溶液。
[0115]
将三聚卤氰与有机溶剂混合,搅拌并至三聚卤氰完全溶解,得到第二溶液。第二溶液所用有机溶剂选自丙酮、n,n-二甲基甲酰胺 (dmf)、n-甲基吡咯烷酮(nmp)、四氢呋喃(thf)和1,4-二氧六环中的一种或者多种。优选地,第二溶液所用有机溶剂为由丙酮和1,4-二氧六环组成的混合物。在该混合物中,丙酮和1,4-二氧六环的重量比为3~4:3~4,优选为3~3.5:3~4,更优选为3~3.2:3.5~ 4。根据本发明的一个实施方式,第二溶液所用有机溶剂为由重量比为3:4的丙酮和1,4-二氧六环组成的混合物。
[0116]
将第一溶液滴加至第二溶液中反应,得到反应产物。所述的二元胺与三聚卤氰的摩尔比可以为3.5~7.5,优选为3.7~6,更优选为 4~5。这样有利于提高超支化聚合物的收率。
[0117]
根据本发明的一个实施方式,首先将第一溶液降温至15~ 40℃,优选为20~35℃,更优选为20~30℃;然后再滴加第一溶液。
[0118]
滴加过程中,将反应体系的温度控制在15~40℃,优选为20~ 35℃。这样有利于提高超支化聚合物的收率。滴加过程中,会明显看到有白色沉淀物大量析出,并伴随着大量的气泡。
[0119]
第一溶液滴加完毕后,继续在15~40℃下反应5~12h。优选地,继续在20~35℃下反应5~10h。
[0120]
将反应产物在6000~10000rpm的转速下离心分离5~15min得到固体产物,将固体产物依次用水和丙酮淋洗,然后真空干燥,得到超支化聚合物改性剂。
[0121]
在本发明中,离心的转速可以为6000~10000rpm,优选为 7000~10000rpm,更优选为8000~10000rpm。离心的时间可以为 5~15min,优选为7~15min,更优选为8~12min。将固体产物依次用水和丙酮淋洗。淋洗用水优选为加热至45~55℃的去离子水,淋洗3~4
次。淋洗用丙酮优选为加热至30~40℃的丙酮,淋洗3~4 次。每次淋洗所用水或丙酮的用量为20~30ml,优选为20~25 ml。将淋洗后的固体真空干燥。真空干燥温度可以为70~95℃,优选为70~85℃,更优选为80~85℃。真空干燥时间可以为7~18h,优选为10~15h。
[0122]
熔融剪切混炼步骤
[0123]
将聚乙醇酸、助剂及超支化聚合物改性剂用混料机混合均匀,得到混合物;将混合物用双螺杆挤出机进行熔融剪切混炼,得到混炼产物。
[0124]
在熔融剪切混炼之前,优选将聚乙醇酸和超支化聚合物在70~ 95℃下真空干燥10~15h,防止其中的水分含量影响聚乙醇酸性能并导致其在复合过程中降解。
[0125]
本发明的双螺杆挤出机包括上腔板、双螺杆和下腔板。双螺杆位于上腔板和下腔板之间,上腔板位于双螺杆的上方,下腔板位于双螺杆的下方。上腔板上设置有加料口,通过加料口将混合物加至双螺杆处挤出。
[0126]
上腔板的温度为220~230℃,优选为220~227℃,更优选为 222~227℃,再优选为222~225℃。
[0127]
下腔板的温度为225~235℃,优选为227~235℃,更优选为 227~232℃,再优选为227~230℃。
[0128]
双螺杆挤出机的螺杆转速为29~50rpm,优选为35~45rpm,更优选为40~43rpm。
[0129]
注塑成型步骤
[0130]
将混炼产物采用注塑机注塑成型,得到聚乙醇酸复合材料。
[0131]
根据本发明的一个实施方式,将混炼产物采用微型注塑机注塑成型,得到聚乙醇酸复合材料。
[0132]
注塑机包括料筒和空腔。料筒用于加入混炼产物。空腔位于料筒的下方,空腔内设置有注塑拉伸样条模具。注塑拉伸样条模具用于将混炼产物形成预定形状的拉伸样条。具体地,将混炼产物加入料筒内,混炼产物从料筒流入注塑拉伸样条模具内,从而形成预定形状的拉伸样条。
[0133]
料筒内温度为200~220℃,优选为205~220℃,更优选为 210~217℃,再优选为215~217℃。
[0134]
注塑拉伸样条模具内的温度为80~160℃,优选为90~150℃,更优选为100~120℃,再优选为105~110℃。
[0135]
根据本发明的一个实施方式,注塑拉伸样条模具内的温度为 85~95℃。根据本发明的另一个实施方式,注塑拉伸样条模具内的温度为145~155℃。
[0136]
在本发明中,熔融剪切混炼步骤和注塑成型步骤为连续的步骤。
[0137]
下面介绍测试方法:
[0138]
断裂伸长率:采用中国台湾高铁gotech电子万能试验机对聚乙醇酸复合材料进行力学拉伸性能测试,测试条件为:温度25℃,拉伸速率1mm/min,样条形状75mm
×
5mm
×
2mm的1ba型哑铃形。每组至少要测试6~8个样条,并确定平均值和标准偏差。根据标准 gb/t 1040.1-2006进行测试。
[0139]
以下实施例和比较例中的原料说明如下:
[0140]
聚乙醇酸:数均分子量为20000~40000,购于内蒙浦景化工公司。
[0141]
制备例1
[0142]
将29.65g 1,3-二氨基丙烷、12.70g的碳酸钠和60ml去离子水混合,在70℃搅拌20min至碳酸钠全部溶解,形成第一溶液。
[0143]
将12.30g三聚氯氰用100ml的溶剂(该溶剂为重量比为3:4的丙酮和1,4-二氧六环组成的混合物)溶解,得到第二溶液。
[0144]
将第一溶液降温至20℃,然后滴加至第二溶液中,滴加完毕, 20℃下反应5h,得到反应产物。
[0145]
将反应产物用高速离心机在转速为8000rpm下离心10min,收集固体产物。将固体产物用去离子水淋洗3次,再用丙酮淋洗3次。每次淋洗溶剂用量为20ml。将淋洗后的固体产物在85℃下真空干燥 12h,得到超支化聚合物改性剂。
[0146]
制备例2
[0147]
与制备例1的区别仅在于,将1,3-二氨基丙烷替换为相应当量的 1,4-二氨基丁烷。
[0148]
制备例3
[0149]
与制备例1的区别仅在于,所用二元胺为58.10g n,n-双(3-氨丙基)甲胺,所用催化剂为12.14g的三乙胺。
[0150]
制备例4
[0151]
与制备例1的区别仅在于,所用二元胺为45.68g反式1,4-环己二胺,所用催化剂为12.14g三乙胺。
[0152]
制备例5
[0153]
与制备例1的区别仅在于,所用二元胺为45.68g 1,4-环己二胺顺反混合物,所用催化剂为9.49g吡啶。
[0154]
制备例6
[0155]
与制备例1的区别仅在于,所用二元胺为43.26g对苯二胺,所用催化剂为9.49g吡啶。
[0156]
制备比较例1
[0157]
与制备例1的区别仅在于,将1,3-二氨基丙烷替换为相应当量的乙二胺。
[0158]
实施例1~9及比较例1~3
[0159]
首先将聚乙醇酸和超支化聚合物在70℃下真空干燥12h。分别按照表1中的用量将聚乙醇酸、超支化聚合物改性剂(各制备例以及制备比较例所制得的)、环氧大豆油及柠檬酸三丁酯在混料机中混合均匀,确保所有的粉体全部均匀附着于聚乙醇酸树脂的表面,得到混合物。
[0160]
将混合物用双螺杆挤出机进行熔融剪切混炼,得到混炼产物;其中,该双螺杆挤出机包括上腔板、双螺杆和下腔板,且双螺杆位于上腔板和下腔板之间,上腔板位于双螺杆的上方,上腔板具有加料口。将混合物通过上腔板的加料口加入至双螺杆处,通过双螺杆挤出。上腔板的温度为225℃,下腔板的温度为227℃;双螺杆挤出机的螺杆转速为40rpm。
[0161]
然后将混炼产物用微型注塑机注塑成哑铃型拉伸样条;其中,微型注塑机包括料筒和空腔。空腔位于料筒的下方,空腔内设置有注塑拉伸样条模具;将混炼产物加入至料筒内,料筒内温度为210℃;混炼产物流至注塑拉伸样条模具(75mm
×
5mm
×
2mm)内,注塑拉伸样条模具内温度为90℃;从而将混炼产物形成75mm
×
5mm
×
2 mm的1ba型哑铃形拉伸样条。
[0162]
采用万能拉伸试验机测定拉伸样条的断裂伸长率。
[0163]
表1
[0164][0165]
表2
[0166]
编号改性剂及其与pga的用量比断裂伸长率/%实施例1制备例1的超支化聚合物;0.0113.84实施例2制备例1的超支化聚合物;0.0315.62实施例3制备例1的超支化聚合物;0.05516.76实施例4制备例1的超支化聚合物;0.0723.76实施例5制备例2的超支化聚合物;0.0715.84实施例6制备例3的超支化聚合物;0.0716.23实施例7制备例4的超支化聚合物;0.0713.87实施例8制备例5的超支化聚合物;0.0719.01实施例9制备例6的超支化聚合物;0.0717.28比较例1制备比较例1的超支化聚合物;0.0710.62比较例208.41比较例3制备例1的超支化聚合物;0.1514.26纯pga05.42
[0167]
由表可知,与纯聚乙醇酸相比,本发明的聚乙醇酸复合材料的断裂伸长率明显提高。由实施例1-4和比较例2、比较例3比较可知,超支化聚合物改性剂需要控制在特定范围内,聚乙醇酸复合材料的韧性改善比较明显;若超支化聚合物改性剂用量过大,则导致聚乙醇酸复合材料的韧性下降;超支化聚合物改性剂用量过小,则无法很好地改善聚乙醇酸复合材料的韧性。由实施例与比较例1比较可知,采用本发明的超支化聚合物作为改性剂时,所得聚乙醇酸复合材料的韧性更好。优选地,采用制备例1的超支化聚合物作为改性剂时,所得聚乙醇酸复合材料的韧性更好。
[0168]
本发明并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员可以想到的任何变形、改进、替换均落入本发明的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
