1.本发明涉及化学原料药的制备技术领域,具体地,涉及一种富马酸沃诺拉赞的制备方法。
背景技术:
2.富马酸沃诺拉赞(vonoprazan fumarate),化学名称为5-(2-氟苯基)-1-(3-吡啶基磺酰基)-3-甲胺甲基-1h-吡咯富马酸盐,由日本武田制药(takeda)研发,于2014年12月在日本首次上市。富马酸沃诺拉赞是一种可逆性质子泵抑制剂,通过抑制k
与h
-k
-atp酶(质子泵)的结合,对胃酸分泌发挥提前终止和强劲、持久的抑制作用,临床上对糜烂性食管炎、幽门螺杆菌感染、十二指肠溃疡及胃溃疡等胃酸相关性疾病具有良好的疗效。此外,富马酸沃诺拉赞还具有相对较高的耐受性和安全性。
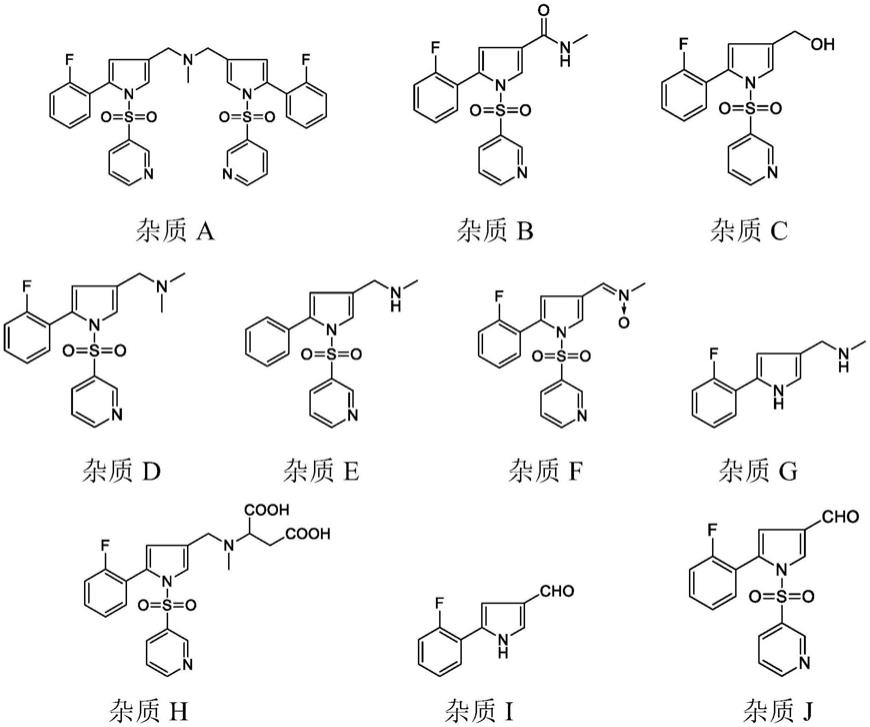
3.沃诺拉赞因合成工艺的不同会产生不同的杂质,zhiqiang luo等报道了通过hplc-uc方法分析富马酸沃诺拉赞的10个杂质,在对富马酸沃诺拉赞的合成工艺进行了全面研究之后,追踪了每个阶段中产生的潜在杂质。其中,杂质-3、4、5、10已在liu等人的研究中被阐述了形成过程。其他杂质可能的形成机理包括,未反应的sma(杂质-8)和中间体2(杂质-9)很可能存在于最终的富马酸沃诺拉赞样品中。此外,杂质-9被认为是沃诺拉赞的氧化降解产物,该降解产物已在降解测试中证实(路线8)。在路线3中,中间体1可以与ch3nh2反应形成杂质-1。此外,杂质-1被推测是沃诺拉赞的基本降解产物,这在强制降解研究中也得到了证实(路线7)。在路线4中,中间体1可与hcl反应形成杂质-7。在路线5中,中间体2可以被nahb4氢化,得到杂质-6。最后,通过在高温下将富马酸沃诺拉赞溶于有机溶剂中进行重结晶,即可得到杂质-2(路线9)。
4.武田公司化合物专利cn200680040789.7公开了以5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛合成富马酸沃诺拉赞的步骤(实施例8),如zhiqiang luo等的报道,将会产生杂质-1、杂质-2、杂质-4、杂质-5、杂质-6、杂质-9等杂质。为降低富马酸沃诺拉赞合成过程中的杂质,尤其是降低以5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛合成富马酸沃诺拉赞的步骤中产生的杂质,专利cn108503621公开了在反应溶液里加入少量吡啶,通过加入吡啶抑制了吡啶磺酰基上吡啶环的部分还原,减少杂质的产生,但需要通过一次重结晶得到单杂小于0.1%,符合药用质量标准的高纯度的富马酸沃诺拉赞。专利cn110734424公开了以草酸盐粗品获得富马酸沃诺拉赞,两步总收率为69.08%,纯度97.72%。专利cn110590746公开了以沃诺拉赞氢溴酸盐制备富马酸沃诺拉赞产品2.35kg,收率为86.71%。hplc纯度99.81%。专利cn107778286以高纯度的n-甲基-1-(3-吡啶磺酰基)-5-(2-氟苯基)-1h-吡咯-3-甲胺盐酸盐制备富马酸沃诺拉赞,纯度达到97%以上。专利cn104926790同样利用高纯度的5-(2-氟苯基)-1-[(吡啶-3-基)磺酰基]-1h-吡咯-3-甲醛,以降低合成中难以去除的杂质,使最终的富马酸沃诺拉赞纯度提升到99.67%。
[0005]
但是上述的各种方法中,加入杂质产生抑制剂、或者改变合成工艺、或者改变中间盐的同时,也引入了新的杂质问题,导致各种杂质普各不相同,公开的杂质情况如下:
[0006][0007]
进一步的,杂质a在工艺中较难控制和去除,杂质h是富马酸沃诺拉赞的降解杂质,是由沃诺拉赞分子结构中的甲胺部分与富马酸分子在加热或强光下发生加成反应产生。现有的合成工艺中为了得到高纯度的富马酸沃诺拉赞,一般都需要在制备得到粗品后选择溶剂进行精制,而精制过程中需要加热溶清,容易产生杂质h,如专利cn201510318995.0中将富马酸沃诺拉赞粗品在异丙醇和二氯甲烷体系中加热回流。
[0008]
综上所述,目前已有大量关于富马酸沃诺拉赞的合成工艺研究,但是现有甲氨基甲基片段的合成工艺存在路线长、总收率低、工艺不稳定、精制过程易产生杂质等不适于工业化生产的缺陷,所以开发一条能够适合工业化生产,无安全环保压力、工艺稳定、杂质控制好的合成路线及方法具有非常重要的意义。
技术实现要素:
[0009]
本发明提供了一种富马酸沃诺拉赞的制备方法,与现有技术相比,该方法杂质含量低、收率较高,无需引入有毒害的反应物,适应工业化生产。
[0010]
本发明针对如背景技术所述的还原过度导致杂质含量提高、胺化试剂不适用工业化的缺陷,对还原剂的选择、甲胺试剂/溶剂的种类、各个反应物的投料量及相关反应参数进行了优化。本发明还优化选择了通过在胺化还原反应后在适当的溶剂中使得游离态沃诺拉赞直接与乙酸形成乙酸盐,并从反应体系中析出,相较于现有技术不成盐形式获得的油状物更容易处理,直接形成固体,使得游离态沃诺拉赞的转化率提高,并可以在同步骤中有效去除杂质,使得后续反应生成富马酸盐的形式更简便。
[0011]
本发明的制备方法为,以5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛为起始物料,经胺化、还原、成盐反应制备得到富马酸沃诺拉赞。
[0012]
本发明的制备包括以下步骤:
[0013]
步骤a、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(vn-1)与甲胺醇溶液、乙酸、还原剂在甲醇中经胺化还原反应再与乙酸成盐生成乙酸沃诺拉赞(vn-2)。
[0014][0015]
步骤b、乙酸沃诺拉赞(vn-2)先解离成游离的沃诺拉赞,再在溶剂中与富马酸成盐生成富马酸沃诺拉赞(vn-3)。
[0016][0017]
作为一种实施方式,步骤a中甲胺醇溶液选自甲胺甲醇溶液、甲胺乙醇溶液、甲胺异丙醇溶液中的任一种,优选甲胺乙醇溶液。
[0018]
作为一种实施方式,步骤a中的甲胺醇溶液中甲胺的含量为30-33%。
[0019]
作为一种实施方式,步骤a中vn-1和甲胺醇溶液中甲胺的摩尔比为1:2-5,优选1:3。
[0020]
作为一种实施方式,步骤a中的还原剂选自三乙酰氧基硼氢化钠、氰基硼氢化钠、三丙酰氧基硼氢化钠、醋酸硼氢化钠中的任一种,优选三乙酰氧基硼氢化钠。
[0021]
作为一种实施方式,步骤a中的vn-1和胺化还原反应中乙酸的摩尔比为1:2-5,优选1:3。
[0022]
作为一种实施方式,步骤a中vn-1和还原剂的摩尔比为1:3-5,优选1:4。
[0023]
作为一种实施方式,步骤a中vn-1和甲醇的质量体积比为1:5-15,优选1:8。
[0024]
作为一种实施方式,步骤b中vn-2与富马酸的投料摩尔比为1:1-1.3,优选1:1.1。
[0025]
作为一种实施方式,步骤b中的溶剂选自甲醇、乙醇、乙酸乙酯中的任一种,优选甲醇。
[0026]
通过本发明所述的方法制备得到的富马酸沃诺拉赞,杂质a的含量低于0.16%,杂质h的含量低于0.06%。
[0027]
本发明的有益效果在于:
[0028]
1.避免使用有刺激性气味及危险性高的甲胺气体,而使用了易于保存的甲胺醇溶液进行反应,适合大批量工艺化生产。
[0029]
2.还原胺化反应时体系中加入了乙酸,提高了反应转化率,提高整体的收率。
[0030]
3.现有技术中游离态的1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基]-n-甲基甲胺不易得到固体,本发明通过在乙酸乙酯溶液中使得游离态与乙酸形成乙酸盐的形式从体系中析出,既可以有效去除杂质,又可以得到固体,使得后续成富马酸盐的形式较为简便。
[0031]
4.由于成乙酸盐过程中有效的去除了杂质,直接成富马酸盐后纯度已达到99%以上,两步均无需再进行精制,同时成富马酸盐时温度低于20℃,因此能很好的避免产生降解杂质h。
[0032]
5.本发明各个步骤的反应在公斤级放量生产中,依然可以保持工艺的可控性,整体的控制反应中杂质的生成,本发明的各步骤稳定性好,保证了富马酸沃诺拉赞的成品质量,总摩尔收率可达75%以上,本案例所有实施例纯度均在99%以上。
附图说明
[0033]
图1为本发明实施例1得到的乙酸沃诺拉赞的hplc图。
[0034]
图2为本发明实施例6得到的富马酸沃诺拉赞的hplc图。
具体实施方式
[0035]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此;本发明实施例中未注明具体条件的实验方法,通常为常规条件,或按照原料或商品制造厂商所建议的条件。未注明来源的试剂,通常为通过商业途径可购得的常规试剂。
[0036]
本发明的起始物料5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛,可以是商业购得的,也可以是通过us20110306769所述的制备方法获得的;其反应式如下:
[0037][0038]
实施例1
[0039]
20℃的条件下,向1l三口瓶中加入甲醇(400ml)、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(50g,150.15mmol)和33%甲胺乙醇(42.75g,450.45mmol)溶液,搅拌1小时后,控温20℃滴加乙酸(45.5g,450.45mmol)。降温,控温5℃加入三乙酰氧基硼氢化钠(36.4g,600.6mmol)。保温搅拌2小时。撤去冷却装置,升温至20℃,搅拌1小时。减压浓缩去除溶剂。向浓缩液中加入纯化水(300ml),搅拌至溶清后,滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,再加入乙酸乙酯,搅拌溶清后降温至5
℃,加入乙酸(10g,166.5mmol),保温搅拌2.5小时。过滤,真空干燥得到类白色结晶性粉末(50.65g,摩尔收率:82.5%)。有关物质中杂质a0.08%,杂质b未检出,杂质c未检出,杂质d0.02%,杂质e0.01%,杂质f未检出,杂质g未检出,杂质h未检出,杂质i未检出,杂质j未检出,纯度99.78%。
[0040]
实施例2
[0041]
20℃的条件下,向250ml三口瓶中加入甲醇(80ml)、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(10g,30.3mmol)和33%甲胺乙醇(5.7g,60.6mmol)溶液,搅拌1小时后,控温20℃滴加乙酸(3.64g,60.6mmol)。降温,控温5℃加入三乙酰氧基硼氢化钠(25.69g,121.2mmol)。保温搅拌2小时。撤去冷却装置,升温至20℃,搅拌1小时。减压浓缩去除溶剂。向浓缩液中加入纯化水(60ml),搅拌至溶清后,滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,再加入乙酸乙酯,搅拌溶清后降温至5℃,加入乙酸(2g,33.3mmol),保温搅拌2.5小时。过滤,真空干燥得到类白色结晶性粉末(9.42g,摩尔收率:76.7%)。有关物质中杂质a0.06%,杂质b未检出,杂质c未检出,杂质d0.02%,杂质e未检出,杂质f未检出,杂质g0.03%,杂质h未检出,杂质i未检出,杂质j0.07%,纯度99.64%。
[0042]
实施例3
[0043]
20℃的条件下,向250ml三口瓶中加入甲醇(80ml)、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(10g,30.3mmol)和33%甲胺乙醇(14.25g,151.5mmol)溶液,搅拌1小时后,控温20℃滴加乙酸(9.1g,151.5mmol)。降温,控温5℃加入三乙酰氧基硼氢化钠(25.69g,121.2mmol)。保温搅拌2小时。撤去冷却装置,升温至20℃,搅拌1小时。减压浓缩去除溶剂。向浓缩液中加入纯化水(60ml),搅拌至溶清后,滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,再加入乙酸乙酯,搅拌溶清后降温至5℃,加入乙酸(2g,33.3mmol),保温搅拌2.5小时。过滤,真空干燥得到类白色结晶性粉末(9.8g,摩尔收率:79.8%)。有关物质中杂质a0.16%,杂质b未检出,杂质c未检出,杂质d0.05%,杂质e0.03%,杂质f未检出,杂质g0.1%,杂质h未检出,杂质i未检出,杂质j未检出,纯度99.40%。
[0044]
实施例4
[0045]
20℃的条件下,向250ml三口瓶中加入甲醇(150ml)、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(10g,30.3mmol)和33%甲胺乙醇(8.55g,90.9mmol)溶液,搅拌1小时后,控温20℃滴加乙酸(5.46g,90.9mmol)。降温,控温5℃加入三乙酰氧基硼氢化钠(19.27g,90.9mmol)。保温搅拌2小时。撤去冷却装置,升温至20℃,搅拌1小时。减压浓缩去除溶剂。向浓缩液中加入纯化水(60ml),搅拌至溶清后,滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,再加入乙酸乙酯,搅拌溶清后降温至5℃,加入乙酸(2g,33.3mmol),保温搅拌2.5小时。过滤,真空干燥得到类白色结晶性粉末(8.94g,摩尔收率:72.8%)。有关物质中杂质a0.06%,杂质b未检出,杂质c未检出,杂质d0.02%,杂质e0.01%,杂质f未检出,杂质g未检出,杂质h未检出,杂质i未检出,杂质j0.14%,纯度
99.65%。
[0046]
实施例5
[0047]
20℃的条件下,向250ml三口瓶中加入甲醇(150ml)、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(10g,30.3mmol)和33%甲胺乙醇(8.55g,90.9mmol)溶液,搅拌1小时后,控温20℃滴加乙酸(5.46g,90.9mmol)。降温,控温5℃加入三乙酰氧基硼氢化钠(32.12g,151.5mmol)。保温搅拌2小时。撤去冷却装置,升温至20℃,搅拌1小时。减压浓缩去除溶剂。向浓缩液中加入纯化水(60ml),搅拌至溶清后,滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,再加入乙酸乙酯,搅拌溶清后降温至5℃,加入乙酸(2g,33.3mmol),保温搅拌2.5小时。过滤,真空干燥得到类白色结晶性粉末(10.02g,摩尔收率:81.6%)。有关物质中杂质a0.12%,杂质b未检出,杂质c0.05%,杂质d0.02%,杂质e0.08%,杂质f未检出,杂质g0.06%,杂质h未检出,杂质i未检出,杂质j未检出,纯度99.54%。
[0048]
实施例6
[0049]
20℃的条件下,向250ml三口瓶中加入纯水(60ml)和10g实施例1中的vn-2(24.7mmol),搅拌至溶清。滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,加入甲醇(20ml),20℃下搅拌溶清后加入富马酸(3.16g,27.2mmol)和甲醇(60ml)的溶液,搅拌30分钟。冷却降温至0℃,搅拌析晶2小时。过滤,真空干燥得到类白色结晶性粉末(10.56g,摩尔收率:94.6%)。有关物质中杂质a0.03%,杂质b未检出,杂质c未检出,杂质d0.01%,杂质e未检出,杂质f未检出,杂质g未检出,杂质h0.02%,杂质i未检出,杂质j未检出,纯度99.91%。
[0050]
实施例7
[0051]
20℃的条件下,向250ml三口瓶中加入纯水(60ml)和10g实施例1中的vn-2(24.7mmol),搅拌至溶清。滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,加入甲醇(20ml),20℃下搅拌溶清后加入富马酸(2.87g,24.7mmol)和甲醇(60ml)的溶液,搅拌30分钟。冷却降温至0℃,搅拌析晶2小时。过滤,真空干燥得到类白色结晶性粉末(9.65g,摩尔收率:86.4%)。有关物质中杂质a0.03%,杂质b未检出,杂质c未检出,杂质d未检出,杂质e未检出,杂质f未检出,杂质g未检出,杂质h0.02%,杂质i未检出,杂质j未检出,纯度99.92%。
[0052]
实施例8
[0053]
20℃的条件下,向250ml三口瓶中加入纯水(60ml)和10g实施例1中的vn-2(24.7mmol),搅拌至溶清。滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,加入甲醇(20ml),20℃下搅拌溶清后加入富马酸(3.73g,32.1mmol)和甲醇(60ml)的溶液,搅拌30分钟。冷却降温至0℃,搅拌析晶2小时。过滤,真空干燥得到类白色结晶性粉末(10.6g,摩尔收率:95.0%)。有关物质中杂质a0.03%,杂质b未检出,杂质c未检出,杂质d0.01%,杂质e未检出,杂质f未检出,杂质g未检出,杂质h0.06%,杂质i未检出,杂
质j未检出,纯度99.87%。
[0054]
对比例:
[0055]
20℃的条件下,向250ml三口瓶中加入甲醇(80ml)、5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-甲醛(10g,30.3mmol)和33%甲胺醇(5.7g,60.6mmol)溶液,搅拌1小时后,控温20℃滴加甲酸(2.80g,60.0mmol)。降温,控温5℃加入三乙酰氧基硼氢化钠(25.69g,121.2mmol)。保温搅拌2小时。撤去冷却装置,升温至20℃,搅拌1小时。减压浓缩去除溶剂。向浓缩液中加入纯化水(60ml),搅拌至溶清后,滴加氨水调节ph值至9~10。加入乙酸乙酯,搅拌、萃取分液。有机相分别用5%碳酸氢钠水溶液、饱和食盐水洗涤一次,再用无水硫酸钠干燥30分钟,过滤。减压浓缩至干后,再加入乙酸乙酯,搅拌溶清后降温至5℃,加入甲酸(1.5g,33.1mmol),保温搅拌2.5小时。过滤,真空干燥得到类白色结晶性粉末(9.80g,摩尔收率:79.7%)。有关物质中杂质a0.17%,杂质b0.02%,杂质c未检出,杂质d0.02%,杂质e0.06%,杂质f未检出,杂质g0.33%,杂质h0.05%,杂质i未检出,杂质j0.01%,纯度99.34%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。