肠促胰岛素类似物的制备方法
1.本发明一般性涉及生物学、化学和医学,更具体地,本发明涉及通过混合的液固相合成(hybrid liquid solid phase synthesis)(hlsps)来合成肠促胰岛素类似物的方法,所述肠促胰岛素类似物在葡萄糖依赖性促胰岛素多肽(gip)、胰高血糖素样肽-1(glp-1)和胰高血糖素(gcg)受体中的一种或多种上具有活性。
2.在过去几十年中,糖尿病的患病率持续上升。2型糖尿病(t2dm)是最常见的糖尿病形式,约占所有糖尿病的90%。t2dm的特点是胰岛素抵抗导致的高血糖水平。目前t2dm的护理标准包括节食和锻炼,以及使用口服降糖疗法和/或注射降糖疗法进行治疗,包括基于肠促胰岛素的疗法,如glp-1受体激动剂、gip/glp-1双受体激动剂,甚至gip/glp-1/gcg(ggg)三受体激动剂。
3.国际专利申请公开号wo 2019/125938和2019/125929一般性描述作为ggg三受体激动剂的肠促胰岛素类似物以及通过标准固相肽合成来合成其的方法。还参见国际专利申请公开号wo 2014/049610、2015/067716、2016/198624、2017/116204、2017/153575和2018/100135。同样,国际专利申请公开号wo2013/164483和2016/111971描述了声明具有glp-1和gip活性的化合物。此外,国际专利申请公开号wo2020/023386描述了具有gip和glp1受体激动剂活性的肽。
4.然而,需要制备这种肠促胰岛素类似物及其中间体的替代方法,以实现具有商业期望纯度的药学上巧妙的生产方式。同样,需要有效的方法和稳定的中间体,且纯化步骤更少,以有效地提供肠促胰岛素类似物。
5.为了满足这一需要,本公开物描述了通过hlsps或天然化学连接(ncl)制备肠促胰岛素类似物的方法,其中此类方法使用二到四种中间体化合物来制备肠促胰岛素类似物。
6.在第一个实施方案中,肠促胰岛素类似物或其可药用盐可包括以下氨基酸序列:
7.yx2qgtftsdysix
13
ldkx
17
ax
19
x
20
afieyllx
28
x
29
gpssx
34
appps,
8.其中x2是aib,x
13
是l或αmel,x
17
是任何具有可用于偶联的官能团的氨基酸,且该官能团与c
16-c
22
脂肪酸偶联,x
19
是q或a,x
20
是aib、αmek、q或h,x
28
是e或a,x
29
是g或aib,x
34
是g或aib(seq id no:4),且c-末端氨基酸任选被酰胺化。在某些实例中,该肠促胰岛素类似物或其可药用盐可具有氨基酸序列:
9.y(aib)qgtftsdysi(αmel)ldkkaq(aib)afieylleggpssgappps(seq id no:5),其中c-末端氨基酸任选被酰胺化。
10.在某些实例中,所述c
16-c
22
脂肪酸可通过具有以下结构的连接基团(linker)连接到肠促胰岛素类似物:
11.(2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)
a-(γglu)
b-co-(ch2)
c-co2h,其中a可以是0、1或2,b可以是1或2,且c可以是16或18。
[0012]
在具体的实例中,该肠促胰岛素类似物或其可药用盐可具有以下氨基酸序列:
[0013]
y(aib)qgtftsdysi(αmel)ldkk((2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)-(γglu)-co-(ch2)
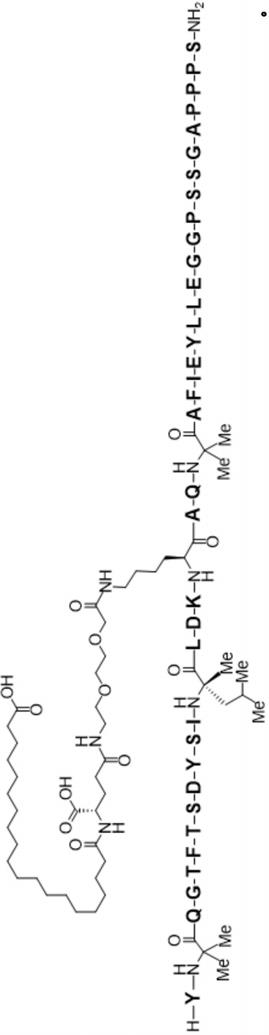
18-co2h)aq(aib)afieylleggpssgappps-nh2(seq id no:6),其可以如以下结构描述:
[0014][0015]
关于通过hlsps制备seq id no:6的肠促胰岛素类似物或其可药用盐的方法,所述方法可包括至少一个偶联四种中间体化合物的步骤,其中所述化合物具有如seq id nos:7、8、9和10中所述的结构。
[0016]
或者,所述方法可包括至少一个偶联四种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:7、11、12和10所述的结构。
[0017]
或者,所述方法可包括至少一个偶联四种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:7、13、14和10所述的结构。
[0018]
在其他实例中,所述方法可包括至少一个偶联三种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:7、13和15所述的结构。
[0019]
或者,所述方法可包括至少一个偶联三种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:16、17和10所述的结构。
[0020]
或者,所述方法可包括至少一个偶联三种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:18、12和10所述的结构。
[0021]
或者,所述方法可包括至少一个偶联三种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:7、45和10所述的结构。
[0022]
或者,所述方法可包括至少一个偶联三种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:7、11和20所述的结构。
[0023]
在其他实例中,所述方法可包括至少一个偶联两种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:19和15所述的结构。
[0024]
或者,所述方法可包括至少一个偶联两种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:18和20所述的结构。
[0025]
上述方法还可以包括在所述偶联步骤之前合成所述两种到四种中间体化合物的步骤。
[0026]
在上述方法中,中间体化合物因此可互相化学偶联或酶促偶联以获得seq id no:6的肠促胰岛素类似物。
[0027]
在上述方法中,所述c
16-c
22
脂肪酸部分和任选的连接基团可在各种中间体化合物偶联之前连接至一种中间体化合物(即,酰化可在完整的肠促胰岛素类似物合成之前发生)。或者,脂肪酸部分可在各种中间体化合物偶联后连接至肠促胰岛素类似物(即,酰化可在肠促胰岛素类似物完全合成后发生)。例如,所述方法可包括至少一个偶联两种中间体化合物的步骤,其中所述化合物或其可药用盐具有如seq id nos:21和18所述的结构,然后偶联具有以下结构的脂肪酸部分:
[0028][0029]
或者,所述方法可包括至少一个偶联两种中间体化合物的步骤,其中所述化合物具有如seq id nos:22和19所述的结构,然后偶联具有以下结构的脂肪酸部分:
[0030][0031]
除上述外,也可以使用ncl制备seq id no:6的肠促胰岛素类似物,其中所述方法可包括至少一个偶联两种中间体化合物的步骤,其中所述化合物可具有选自下述的结构:
[0032]
seq id nos:23和24,
[0033]
seq id nos:39和24,
[0034]
seq id nos:25和26,
[0035]
seq id nos:40和26,和
[0036]
seq id nos:27和26。
[0037]
在另一项实施方案中,肠促胰岛素类似物或其可药用盐可以包含以下氨基酸序列:
[0038]
y(aib)egt(αmef(2f))tsd(4pal)si(αmel)ld(orn)k((2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)
2-(γ-glu)-co-(ch2)
16-co2h)aq(aib)efi(d-glu)(αmey)lieggpssgappps-nh2(seq id no:29),其可以如以下结构描述:
[0039][0040]
关于通过hlsps制备seq id no:29的肠促胰岛素类似物或其可药用盐的方法,所述方法可包括将以下中间体化合物中的至少一种偶联至另一种中间体化合物的至少一个
步骤,其中所述化合物具有seq id nos:30、31、32、34、35、36和/或37中所述的结构。
[0041]
本发明提供了制备seq id no:29的肠促胰岛素类似物的方法,该方法包括以下步骤:
[0042]
通过混合的液固相合成,偶联从以下组中选择的中间体化合物:
[0043]
a.seq id nos:7、62、42和31,
[0044]
b.seq id nos:43和44。
[0045]
除上述方法外,本文中的实施方案还包括中间体化合物本身(例如,seq id nos:7-28和30-41)以及包含它们的组合物。
[0046]
本文中类似物的优点是,它们可以用作糖尿病、血脂异常、非酒精性脂肪性肝病(nafld)、代谢综合征、非酒精性脂肪性肝炎(nash)和肥胖症的有效治疗,以及与glp-1和/或gip和/或胰高血糖素的调节相关的其他疾病或病症。
[0047]
本文所述方法的优点包括若干工艺改进,例如,最初通过spps制备的较短片段能够通过hlsps普遍提高纯度和提升产率。
[0048]
本文所述方法的优点包括,在spps中偶联的效率不仅取决于化学转化中涉及的实际残基,而且还受连接在树脂上的结构的影响(即,对于某些序列,溶解度/聚集问题是众所周知的)。采用较短片段,更灵活的路径可用于复杂氨基酸残基的偶联,并且能够重新设计片段结构以解决更困难的转化。
[0049]
本文所述方法的优点包括在合成过程中对杂质的改进的控制策略,其可使粗肽的最终杂质谱得到改善,并简化/减少色谱负担,从而节省成本。
[0050]
本文所述方法的优点包括,通过spps合成较短片段可以减少洗涤循环,减少试剂体积,并使用更绿色的溶剂,从而降低过程质量强度(pmi)。
[0051]
本文所述方法的优点包括使用较短片段,可显著降低长分子线性构建中典型的失败风险。
[0052]
本文所述方法的优点包括,液相和固相合成的组合更适合于新的生产平台并引入其他创新技术。
[0053]
本文所述方法的优点包括,通过使用多个独立片段而导致制备过程的供应链和物流的灵活性。
[0054]
本文所述方法的优点包括,使用片段的并行制备可以通过片段的并行处理提供减少的制备循环。
[0055]
本文所述方法的优点包括当前良好制造规范(cgmp)的汇集步骤可以在标准设施中进行,而无需专用设备。
[0056]
除非另有定义,否则本文中使用的所有技术和科学术语具有与本发明所属领域的技术人员通常理解的相同含义。尽管在肠促胰岛素类似物、药物组合物和方法的实践或测试中可以使用与本文所述类似或等效的任何方法和材料,但本文描述了优选的方法和材料。
[0057]
此外,不定冠词“一个”或“一种”提及一个要素时并不排除存在不止一个要素的可能性,除非上下文明确要求有且仅有一个要素。因此,不定冠词“一个”或“一种”通常意味着“至少一个”。
[0058]
缩写和定义
[0059]
某些缩写的定义如下:“aeea”是指2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基,“d-glu”或“e”是指d-谷氨酸,氨基酸序列中的“e”是指d-谷氨酸,“aib”是指α-氨基异丁酸,“αmel”是指α-甲基亮氨酸,“αmek”是指α-甲基赖氨酸,“boc”是指叔丁氧基羰基,“bu”是指丁基,“t-bu”是指叔丁基,“ctc”是指氯三苯甲基氯,“dcm”是指二氯甲烷,“dic”是指二异丙基碳二亚胺,“dmf”是指二甲基甲酰胺,“dmso”是指二甲基亚砜,“dtt”是指二硫苏糖醇,“edta”是指乙二胺四乙酸,“fmoc”是指芴基甲氧基羰基氯,“hr”是指小时,“ipa”是指异丙醇,“ipac”是指乙酸异丙酯,“min”是指分钟,“me”是指甲基,“mtbe”是指甲基叔丁基醚,“oxyma”是指氰基羟基亚氨基乙酸乙酯,“pg”是指保护基团,“pip”是指哌啶,“spps”是指固相肽合成,“tfa”是指三氟乙酸,“tips”是指三异丙基硅烷,且“trt”是指三苯甲基。
[0060]
如本文所用,“约”是指在一个或多个值的统计学意义范围内,例如,所述浓度、长度、分子量、ph、序列同一性、时间量级、温度或体积。所述值或范围可以在一个数量级内,通常在给定值或范围的20%内,更通常在10%内,甚至更通常在5%内。“约”所包含的允许变化将取决于所研究的特定系统,并且本领域技术人员可以容易地理解。
[0061]
如本文所用,并涉及gip、glp-1或gcg受体中的一个或多个,“活性”、“活化”、“激活”等指化合物(如本文所述的肠促胰岛素类似物)结合并诱导受体响应的能力,如使用本领域已知的试验所测得,如下文所述的体外试验。
[0062]
如本文所用,“具有可用于偶联的官能团的氨基酸”指任何具有可通过例如连接基团与脂肪酸偶联的官能团的天然或非天然氨基酸。此类官能团的实例包括但不限于炔基、烯基、氨基、叠氮基、溴、羧基、氯、碘和硫醇基。包括此类官能团的天然氨基酸的实例包括lys/k(氨基)、cys/c(硫醇)、glu/e(羧基)和asp/d(羧基)。
[0063]
如本文所用,“类似物”指激活靶受体并引发至少一种由该受体的天然激动剂引发的体内或体外效应的化合物,例如合成的肽或多肽。
[0064]
如本文所用,“c
16-c
22
脂肪酸”指具有16至22个碳原子的羧酸。适用于本文的c
16-c
22
脂肪酸可以是饱和单酸或饱和二酸。如本文所用,“饱和”指脂肪酸不含碳-碳双键或叁键。
[0065]
如本文所用,“双受体活性”是指在gip、glp-1和gcg受体中的一个或多个上具有激动剂活性的肠促胰岛素类似物,尤其是在一个或多个受体上具有平衡且足够活性的类似物,以提供该受体激动的益处,同时避免了与过多活性相关的不必要的副作用。此外,具有双受体活性的肠促胰岛素类似物在gip、glp-1和gcg受体中的一个或多个上具有延长的作用持续时间,这有利地允许每天一次、每周三次、每周两次或每周一次给药。
[0066]
如本文所用,“葡萄糖依赖性促胰岛素多肽”或“gip”是指在葡萄糖存在的情况下,通过刺激胰腺β细胞的胰岛素分泌而在葡萄糖稳态中发挥生理作用的肽,尤其是人gip(seq id no:1)。
[0067]
如本文所用,“胰高血糖素样肽-1”或“glp-1”指刺激葡萄糖依赖性的胰岛素分泌的肽,并已被证明可预防糖尿病患者的高血糖症,尤其是人glp-1(seq id no:2)。
[0068]
如本文所用,“胰高血糖素”或“gcg”指通过结合并激活肝细胞上的胰高血糖素受体帮助维持血糖的肽,其使肝脏通过称为糖原分解的过程释放(以糖原的形式存储的)葡萄糖,尤其是人gcg(seq id no:3)。
[0069]
如本文所用,“肠促胰岛素类似物”是指与gip、glp-1和gcg各自,尤其是人gip(seq id no:1)、人glp-1(seq id no:2)和人gcg(seq id no:3)具有结构相似性但存在多处差异
的化合物。本文所述的肠促胰岛素类似物包括氨基酸序列,其导致化合物对gip、glp-1和gcg受体中的一个或多个具有亲和力和活性(即,双重激动剂活性或三重激动剂活性)。
[0070]
如本文所用,“可药用缓冲液”指本领域技术人员已知的任何标准的药物缓冲液。
[0071]
如本文所用,“三受体活性”是指具有gip、glp-1和gcg受体激动剂活性的肠促胰岛素类似物,尤其是在受体处具有平衡且足够活性的类似物,以提供受体激动的益处,同时避免了与过多活性相关的不期望的副作用。此外,具有三受体活性的肠促胰岛素类似物在gip、glp-1和gcg受体中的一个或多个上具有延长的作用持续时间,这有利地允许每天一次、每周三次、每周两次或每周一次给药。
[0072]
组合物
[0073]
本文中肠促胰岛素类似物的结构特征导致化合物在gip、glp-1和gcg受体的一个或多个上具有足够的活性,以获得在一个或多个受体上的活性的有利效果(即,双受体活性或三受体活性),但在任何一个受体上的活性都不会过多以至于压倒在其他两个受体上的活性或者当以足以导致全部三个受体的活性的剂量施用时产生不期望的副作用。
[0074]
本文中肠促胰岛素类似物的结构特征还使得化合物具有与作为治疗性处理的可开发性相关的许多其他有利属性,包括改善所述类似物在水溶液中的溶解度、改善化学和物理制剂稳定性、延长药代动力学曲线,以及最大限度地降低免疫原性的可能性。
[0075]
应当注意,前文罗列的结构特征是示例性的,并不全面,并且本文所述示例性类似物的有利特征的组合不是任何单独修饰的结果,而是通过本文所述结构特征的新颖组合来实现。此外,前述修饰列表的上述效果并非排他性的,因为许多这些修饰还具有对本文所述化合物的特性重要的其他效果,如下文所述。
[0076]
本文中肠促胰岛素类似物的氨基酸序列包含天然存在的氨基酸,通常在本文中使用标准的一个或三个字母代码描述(例如,l/leu=亮氨酸),以及天然氨基酸的α-甲基取代的残基(例如,(αmel、αmek、αmey、αmef(2f))和某些其他非天然氨基酸,如aib、鸟氨酸、4-pal。这些氨基酸的结构如下所示:
[0077]
[0078]
如上所述,本文的肠促胰岛素类似物包括例如通过连接基团与具有可用于偶联的官能团的天然或非天然氨基酸偶联的脂肪酸部分。这种偶联有时被称为酰化。在某些情况下,具有可用于偶联的官能团的氨基酸可以是k、c、e和d,尤其是seq id no:5或seq id no:29中17位的k,其中偶联是连接到k侧链的ε-氨基。脂肪酸部分充当白蛋白粘合剂,并提供产生长效化合物的潜力。
[0079]
本文的肠促胰岛素类似物利用c
16-c
22
脂肪酸,其通过直键或连接基团与氨基酸的官能团化学偶联。脂肪酸的长度和组成影响肠促胰岛素类似物的半衰期、其在体内动物模型中的效能以及它们的溶解度和稳定性。与c
16-c
22
饱和脂肪族一元酸或二元酸的偶联产生肠促胰岛素类似物,其在体内动物模型中表现出期望的半衰期、期望的效能以及期望的溶解度和稳定性特征。
[0080]
本文所用的饱和c
16-c
22
脂肪酸的实例包括但不限于棕榈酸(十六烷酸)(c
16
一元酸)、十六烷二酸(c
16
二元酸)、珍珠酸(十七烷酸)(c
17
一元酸)、十七烷二酸(c
17
二元酸)、硬脂酸(c
18
一元酸)、十八烷二酸(c
18
二元酸)、十九烷酸(十九碳酸)(c
19
一元酸)、十九烷二酸(c
19
二元酸)、二十烷酸(花生酸)(c
20
一元酸)、二十烷二酸(c
20
二元酸)、二十一烷酸(二十一碳酸)(c
21
一元酸)、二十一烷二酸(c
21
二元酸)、二十二烷酸(山萮酸)(c
22
一元酸)、二十二烷二酸(c
22
二元酸),包含其支链的和被取代的衍生物。
[0081]
在某些情况下,c
16-c
22
脂肪酸可以是饱和c
18
一元酸、饱和c
18
二元酸、饱和c
19
一元酸、饱和c
19
二元酸、饱和c
20
一元酸、饱和c
20
二元酸,以及其支链的和被取代的衍生物。
[0082]
在某些情况下,连接基团可具有1至4个氨基酸、氨基聚乙二醇羧酸酯或其混合物。在某些情况下,氨基聚乙二醇羧酸酯具有以下结构:
[0083]
h-{nh-ch
2-ch
2-[o-ch
2-ch2]
m-o-(ch2)
p-co}
n-oh,
[0084]
其中m是1-12的任意整数,n是1-12的任意整数,且p是1或2。
[0085]
在某些情况下,连接基团可具有一个或多个(2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)部分,以及任选的1-4个氨基酸。
[0086]
在连接基团包含至少一种氨基酸的情况下,该氨基酸可以是1-4个glu或γglu氨基酸残基。在某些情况中,连接基团可包含一个或两个glu或γglu氨基酸残基,包括其d-形式。例如,连接基团可包含一个或两个γ-glu氨基酸残基。或者,连接基团可包含1-4个氨基酸残基(例如glu或γglu氨基酸),其用于与至多36个(2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)部分组合。尤其是,所述连接基团可以是1-4个glu或γglu氨基酸和1-4个(2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)部分的组合。在其他情况下,所述连接基团可以是1或2个γglu氨基酸和1或2个(2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)部分的组合。
[0087]
在某些情况下,本文所述的肠促胰岛素类似物包括具有下式结构的连接基团和脂肪酸组分:
[0088]
(2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)
a-(γglu)
b-co-(ch2)
c-co2h,
[0089]
其中a是0、1或2,b是1或2,且c是16或18。
[0090]
在一个具体实例中,a是2,b是1,且c是16,其结构如下所述:
[0091]
[0092]
在另一个具体实例中,a是1,b是2,且c是18,其结构如下所述:
[0093][0094]
在另一个具体实例中,a是0,b是2,且c是18,其结构如下所述:
[0095][0096]
在另一个具体实例中,a是1,b是1,且c是18,其结构如下所述:
[0097][0098]
在具体实例中,肠促胰岛素类似物的整体结构是seq id no:6。
[0099]
在具体实例中,肠促胰岛素类似物的整体结构是seq id no:29。
[0100]
本文的肠促胰岛素类似物对gip、glp-1和gcg受体中各自的亲和力可使用本领域已知的用于检测受体结合水平的技术来检测,包括例如下文实施例中描述的那些,并且通常表示为抑制常数(ki)值。本文的肠促胰岛素类似物在一个或多个所述受体上的活性也可使用本领域已知的技术进行检测,包括例如下文所述的体外活性测定,并且通常表示为有效浓度50(ec
50
)值,其是在剂量-效应曲线中引起半最大刺激的化合物浓度。
[0101]
本文的肠促胰岛素类似物可配制为药物组合物,其可通过胃肠外途径(例如,皮下、静脉内、腹膜内、肌肉内或经皮)施用。此类药物组合物及其制备方法为本领域所熟知。参见例如“remington:the science and practice of pharmacy”(troy ed.,lippincott,williams&wilkins第21版,2006)。
[0102]
本文的肠促胰岛素类似物可与多种无机和有机酸/碱中的任何一种反应以形成可药用的酸/碱加成盐。可药用的盐和制备它们的常用技术在本领域是众所周知的(参见例如stahl等人,“handbook of pharmaceutical salts:properties,selection and use”(wiley-vch第二版,2011))。本文所用的可药用盐包括钠盐、三氟乙酸盐、盐酸盐和/或乙酸盐。
[0103]
本公开物还提供并因此包含合成本文所述肠促胰岛素类似物或其可药用盐的新中间体化合物和方法。本文的中间化合物和肠促胰岛素类似物可通过本领域已知的多种技术制备。例如,下文实施例中说明了一种使用标准固相肽合成两种或多种中间体化合物、随后进行其hlsps的方法。所述各个路径的具体合成步骤可以不同方式组合以制备本文的肠促胰岛素类似物。试剂和起始材料对本领域技术人员而言容易获得。
[0104]
本文的肠促胰岛素类似物通常在宽的剂量范围内有效。例如,每周施用一次的剂量可能在约0.01至约30mg/人/周的范围内,在约0.1至约10mg/人/周的范围内,或甚至在约0.1至约3mg/人/周的范围内。因此,本文所述的肠促胰岛素类似物可每日、每周三次、每周两次或每周一次给药,尤其是每周一次施用。
[0105]
本文的肠促胰岛素类似物可用于治疗多种病况、病症、疾病或症状。具体而言,下文提供在个体中治疗t2dm的方法,其中此类方法至少包括向需要此类治疗的个体施用有效量的本文所述肠促胰岛素类似物或其可药用盐的步骤。
[0106]
方法
[0107]
中间体化合物的标准固相肽合成:
[0108]
本文的肠促胰岛素类似物可通过本领域已知的任何数量的标准肽合成方法制备,尤其是spps。spps构建使用标准fmoc肽化学技术完成,采用使用自动肽合成器的序列偶联。spps的方法在本领域中是众所周知的,不需要在本文中详尽描述。一般参见“fmoc solid phase peptide synthesis:a practical approach”(chan&white ed.,oxford university press 2000),和merrifield(1963)j.am.chem.soc.85:2149-2154。
[0109]
对于脱保护,树脂用dmf溶胀,然后用20%pip/dmf(3x30分钟)脱保护。随后的fmoc脱保护使用20%pip/dmf(1x5-20分钟,1x20-30分钟)处理,1x5-20分钟、1x20分钟和1x30分钟的处理顺序可用于更难的脱保护。
[0110]
脱保护后,用5x2min、10体积的dmf洗涤树脂。氨基酸预活化使用dic/oxyma dmf溶液在室温下进行30分钟。活化的氨基酸与树脂结合的肽的偶联对于每个单个氨基酸在特定时间内发生。每次偶联后,使用10体积dmf,用5x2分钟进行溶剂洗涤。
[0111]
为了分离终产物,用10体积的dcm洗涤树脂结合的产物5x2分钟以去除dmf。用2x2min 10体积的ipa洗涤树脂以去除dcm,用10体积的mtbe洗涤5x2min,然后在40℃的真空条件下干燥产物。将该树脂结合的产物冷藏(-20℃)。
[0112]
对于分析,使用按照如下比例的tfa/h2o/tips/dtt的酸性混合物从树脂中切割肽:(0.93v/0.04v/0.03v/0.03w)。用dcm(4-5体积,3x30分钟)将树脂溶胀并倾出。将切割试剂混合物(4-5体积)添加到预溶胀的树脂中,并在室温下搅拌该混悬液2小时。过滤溶液,然后用少量dcm洗涤树脂,并与切割溶液混合。将所得溶液倒入7-10体积的冷(0℃)mtbe中。将混悬液在0℃下老化30分钟,离心所得沉淀,并倒出该澄清溶液。将残余物悬浮于相同体积的mtbe中,所得混悬液再次离心并倒出。倒出后,将沉淀的肽的澄清mtbe溶液在真空中于40℃下干燥过夜。
[0113]
肠促胰岛素类似物的混合的液固相合成:
[0114]
如上所述通过spps制备的中间体化合物可以组合以获得seq id no:6或29的肠促胰岛素类似物。hlsps的方法在本领域中是众所周知的,不需要在本文中详尽描述。一般参见美国专利申请公开号2011/0046349;和albericio等人(1997)methods enzymol.289:313-336,bray等人(2003)nature rev.drug discovery 2:587-593,dalcol等人(1995)j.org.chem.7575-60:7581,gauthier等人(1991)tettrahedron lett.32:577-580,schneider等人(2005)j.peptide sci.11:744-753,smith,organic synthesis(academic press第4版2016),和zhang等人(2008)org.process res.dev.12:101-110。
[0115]
简言之,hlsps涉及独立的中间体化合物合成和化合物偶联。此处应用的制备seq id no:6的肠促胰岛素类似物的方法至少包括偶联以下四种中间体化合物的步骤,其中这些化合物具有seq id nos:7、8、9和10中所述的结构。
[0116]
在某些情况中,所述片段可按以下顺序偶联:seq id no:7至seq id no:8至seq id no:9至seq id no:10(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策
略,所述片段可以以不同的顺序偶联。
[0117]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下四种中间体化合物的步骤,其中这些化合物具有seq id nos:7、11、12和10中所述的结构。
[0118]
在某些情况中,所述片段按以下顺序偶联:seq id no:7至seq id no:11至seq id no:12至seq id no:10(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0119]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下四种中间体化合物的步骤,其中这些化合物具有seq id nos:7、13、14和10中所述的结构。
[0120]
在某些情况中,所述片段按以下顺序偶联:seq id no:7至seq id no:13至seq id no:14至seq id no:10(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0121]
或者,制备seq id no:6的肠促胰岛素类似物的一种方法至少包括偶联以下三种中间体化合物的步骤,其中这些化合物具有seq id nos:7、13和15中所述的结构。
[0122]
在某些情况中,所述片段按以下顺序偶联:seq id no:7至seq id no:13至seq id no:15(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0123]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下三种中间体化合物的步骤,其中这些化合物具有seq id nos:16、17和10中所述的结构。
[0124]
在某些情况中,所述片段按以下顺序偶联:seq id no:16至seq id no:17至seq id no:10(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0125]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下三种中间体化合物的步骤,其中这些化合物具有seq id nos:18、12和10中所述的结构。
[0126]
在某些情况中,所述片段按以下顺序偶联:seq id no:18至seq id no:12至seq id no:10(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0127]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下三种中间体化合物的步骤,其中这些化合物具有seq id nos:7、45和10中所述的结构。
[0128]
在某些情况中,所述片段按以下顺序偶联:seq id no:7至seq id no:45至seq id no:10(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0129]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下三种中间体化合物的步骤,其中这些化合物具有seq id nos:7、11和20中所述的结构。
[0130]
在某些情况中,所述片段按以下顺序偶联:seq id no:7至seq id no:11至seq id no:20(即,从c-末端到n-末端)。在其他情况下,通过适当的保护基团策略,所述片段可以以不同的顺序偶联。
[0131]
或者,制备seq id no:6的肠促胰岛素类似物的一种方法至少包括偶联以下两种中间体化合物的步骤,其中这些化合物具有seq id nos:19和15中所述的结构。
[0132]
制备seq id no:6的肠促胰岛素类似物的另一种方法至少包括偶联以下两种中间
体化合物的步骤,其中这些化合物具有seq id nos:18和20中所述的结构。
[0133]
或者,制备seq id no:6的肠促胰岛素类似物的其他方法使用与上述相同的分开连接,但是却首先偶联骨架的所有氨基酸片段,然后引入脂肪酸侧链部分作为最后的化学转化,随后进行总体脱保护。此时,例如相应的pg可在lys17处实施,其可在存在其他pg(例如,boc、tbu和/或trt)的情况下选择性地去除。在某些情况下,制备seq id no:6的肠促胰岛素类似物的方法至少包括偶联以下中间体化合物的步骤,其中这些化合物具有seq id nos:21和18中所述的结构,以及
[0134][0135]
在某些情况下,制备seq id no:6的肠促胰岛素类似物的方法至少包括偶联以下中间体化合物的步骤,其中这些化合物具有seq id nos:22和19中所述的结构,以及
[0136][0137]
或者,制备seq id no:6的肠促胰岛素类似物的其他方法至少包括通过ncl方法偶联脱保护的化合物中间体(例如硫酯片段和酰胺片段)的步骤。此处,例如ala21可被cys的天然对映异构体替代,并且在完成以下中间体化合物的连接步骤后,seq id no:6可通过将cys脱硫以提供所需的ala21而获得,其中所述中间体化合物具有seq id no:23和24中所述的结构。
[0138]
或者,硫酯(seq id no:23)可被中间体化合物替代,所述中间体化合物中的c末端的-c-p-or酯(cpe)部分可用作被遮蔽的硫酯,以帮助具有seq id no:39和24所述结构的化合物的连接步骤。
[0139]
在其他情况下,可以用cys代替ala18,并在以下中间体化合物的天然化学连接后脱硫,其中此类化合物具有如seq id nos:25和26所述的结构。
[0140]
或者,硫酯(seq id no:25)可被中间体化合物替代,所述中间体化合物中的-cys-pro-or酯(cpe)部分可用作被遮蔽的硫酯,以帮助具有seq id no:40和26所述结构的化合物的连接步骤。
[0141]
或者,制备seq id no:5的肠促胰岛素类似物的其他方法至少包括通过ncl方法偶联以下中间体化合物(例如非酰化硫酯片段和酰胺片段)的步骤,其中此类化合物具有seq id no:27和26中所述的结构。
[0142]
或者,硫酯(seq id no:27)可被中间体化合物替代,所述中间体化合物中的-cys-pro-or酯(cpe)部分可用作被遮蔽的硫酯,以帮助具有seq id no:41和26所述结构的化合物的连接步骤。
[0143]
在另一个实施方案中,seq id no:29可通过偶联seq id no:43和seq id no:44,并随后脱保护以生成seq id no:29来合成。
[0144]
在另一个实施方案中,seq id no:48可通过使用seq id no:20和seq id no:18来合成。将seq id no:48脱保护以生成seq id no:6。
[0145]
在另一个实施方案中,seq id no:53可通过ncl使用seq id no:51和seq id no:52来合成。
[0146]
在另一个实施方案中,seq id no:53可通过ncl使用seq id no:52和seq id no:54来合成。
[0147]
为了有效制备seq id nos:9、12、14、15、17、20、23和25的化合物中间体,以下使用脂肪族侧链
[0148][0149]
和连接有脂肪族侧链的fmoc-l-lys-oh氨基酸
[0150][0151]
来合成。
[0152]
为了提高spps的纯度和效率,可以使用以下二聚体、三聚体和四聚体来制备seq id nos:10、15、20、21、22、23、25和27,其中可以使用氨基酸结构单元通过spps或液相合成来合成以下结构:
[0153][0154][0155]
其它方法/用途:
[0156]
本文的肠促胰岛素类似物可用于多种治疗应用。例如,肠促胰岛素类似物可用于治疗个体肥胖症的方法中,其中此类方法至少包括向需要此类治疗的个体施用有效量的本文所述肠促胰岛素类似物或其可药用盐的步骤。
[0157]
此外,肠促胰岛素类似物可用于诱导个体的非治疗性体重减轻的方法中,其中此类方法至少包括向需要此类治疗的个体施用有效量的本文所述肠促胰岛素类似物或其可药用盐的步骤。
[0158]
此外,本文的肠促胰岛素类似物可用于在个体中治疗代谢综合征的方法中,其中此类方法至少包括向需要此类治疗的个体施用有效量的本文所述肠促胰岛素类似物或其可药用盐的步骤。
[0159]
此外,本文的肠促胰岛素类似物可用于在个体中治疗nash的方法中,其中此类方法至少包括向需要此类治疗的个体施用有效量的本文所述肠促胰岛素类似物或其可药用盐的步骤。
[0160]
此外,本文的肠促胰岛素类似物可用于在个体中治疗nafld的方法中,其中此类方法至少包括向需要此类治疗的个体施用有效量的本文所述肠促胰岛素类似物或其可药用盐的步骤。
[0161]
在这些方法中,可通过例如观察血糖的显著降低、观察胰岛素的显著增加、观察hba1c的显著降低和/或观察体重的显著降低来评估肠促胰岛素类似物的有效性。
[0162]
或者,本文的肠促胰岛素类似物或其可药用盐可用于改善需要其的个体的骨强度。在某些情况下,需要此种治疗的个体具有骨不足(hypo-ostosis)或骨质减少症(hypo-osteoidosis),或者正在从骨折、矫正手术、假体植入、牙齿植入和/或脊柱融合术中愈合。所述肠促胰岛素类似物也可用于治疗其他疾病,如帕金森病或阿尔茨海默症。
实施例
[0163]
提供以下非限定性实施例仅用于举例说明的目的,而非限定其范围。
[0164]
肽和多肽合成
[0165]
实施例1:中间体化合物1的固相肽合成
[0166]
中间体化合物1(seq id no:7)或其可药用盐可通过标准spps合成。简言之,spps使用sieber树脂(负载系数0.6-0.9mmol/g)进行,条件如下表1所示。
[0167]
表1:用于实施例1的spps条件
[0168][0169][0170]
fmoc脱保护、片段切割和分离:用10v 20%哌啶/dmf搅拌sieber树脂上的片段两次20-30分钟,然后用10v dmf洗涤六次。使用10v dcm将sieber树脂上的脱fmoc片段溶胀两次,持续10-20分钟。将装有树脂的反应器冷却至约15℃,并将20v 5%tfa/dcm装入反应器,然后在氮气中在维持约15℃的温度下搅拌2小时。过滤树脂,并用3x10v dcm洗涤。将全部滤液合并在一起。在减压下将dcm从所得溶液中除去,同时保持内部温度≤20℃直至22.5v剩余体积。将mtbe(25v)加入该溶液中,将dcm/mtbe溶剂再次减压除去,同时保持内部温度≤20℃直至22.5v剩余体积。重复加入mtbe/蒸馏操作,直至在上清液中的片段剩余浓度还未达到《0.11重量%。然后,将所得浆体过滤,期间保持温度在约15℃。向该滤饼中加入14v新鲜的mtbe,在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得固体在约35℃下干燥。
[0171]
实施例2:中间体化合物2的固相肽合成
[0172]
中间体化合物2(seq id no:8)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-gly-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表2所示。
[0173]
表2:实施例2的spps条件
[0174][0175][0176]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将10v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和,然后将5v dmso加入该滤液中。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并。将该片段溶液真空浓缩至6-10v,保持温度≤35℃(剩余dcm浓度≤15%)。将该片段的dmso溶液历经2-6小时时间(《1l/min)在约25℃加入11-15v h2o中。将沉淀片段形成的浆体在约25℃搅拌30-40分钟,然后过滤。将所得固体在约25℃悬浮于8-12v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0177]
实施例3:中间体化合物3的固相肽合成
[0178]
中间体化合物3(seq id no:9)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-ala-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表3所示。
[0179]
表3:实施例3的spps条件
[0180][0181][0182]
*fmoc-l-lys(t-buooc-(ch2)
18-coo-γ-l-glu-aeea)
[0183][0184]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v的1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在维持约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤x%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0185]
实施例4:中间体化合物4的固相肽合成
[0186]
中间体化合物4(seq id no:10)或其可药用盐可通过标准spps合成。简言之,spps
使用fmoc-leu-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表4所示。
[0187]
表4:实施例4的spps条件
[0188][0189][0190]
*boc-l-tyr(t-bu)-aib-l-gln(trt)-gly-oh的结构如下:
[0191][0192]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在维持约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入2v dmso,并真空除去剩余的dcm(剩余dcm浓度≤5%),保持温度≤20℃。将该片段的dmso溶液历经2-6小时时间(《1l/min)加入7-9v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0193]
实施例5:中间体化合物5的固相肽合成
[0194]
中间体化合物5(seq id no:11)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-gly-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表5所示。
[0195]
表5:实施例5的spps条件
[0196][0197]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将10v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和,然后将5v dmso加入该滤液中。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并。将片段溶液真空浓缩至6-10v,保持温度≤35℃(剩余dcm浓度≤15%)。将该片段的dmso溶液历经2-6小时时间(《1l/min)在约25℃加入11-15v h2o中。将沉淀片段形成的浆体在约25℃搅拌30-40分钟,然后过滤。将所得固体在约25℃悬浮于8-12v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0198]
实施例6:中间体化合物6的固相肽合成
[0199]
中间体化合物6(seq id no:12)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-aib-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表6所示。
[0200]
表6:实施例6的spps条件
[0201][0202]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3vdcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤15%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0203]
实施例7:中间体化合物7的固相肽合成
[0204]
中间体化合物7(seq id no:13)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-gly-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表7所示。
[0205]
表7:实施例7的spps条件
[0206][0207][0208]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将10v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和,然后将5v dmso加入该滤液中。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并。将片段溶液真空浓缩至6-10v,保持温度≤35℃(剩余dcm浓度≤15%)。将该片段的dmso溶液历经2-6小时时间(《1l/min)在约25℃加入11-15v h2o中。将沉淀片段形成的浆体在约25℃搅拌30-40分钟,然后过滤。将所得固体在约25℃悬浮于8-12v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0209]
实施例8:中间体化合物8的固相肽合成
[0210]
中间体化合物8(seq id no:14)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-ala-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表8所示。
[0211]
表8:实施例8的spps条件
[0212][0213]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在维持约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤15%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0214]
实施例9:中间体化合物9的固相肽合成
[0215]
中间体化合物9(seq id no:15)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-ala-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表9所示。
[0216]
表9:实施例9的spps条件
[0217]
[0218][0219]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在维持约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中
加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤15%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0220]
实施例10:中间体化合物10的固相肽合成
[0221]
中间体化合物10(seq id no:16)或其可药用盐可通过标准spps合成。简言之,spps使用sieber树脂(负载系数0.6-0.9mmol/g)进行,条件如表10所示。
[0222]
表10:实施例10的spps条件
[0223]
[0224][0225]
fmoc脱保护、片段切割和分离:用10v 20%哌啶/dmf搅拌sieber树脂上的片段两次20-30分钟,然后用10v dmf洗涤六次。使用10v dcm将sieber树脂上的脱fmoc片段溶胀两次,持续10-20分钟。将装有树脂的反应器冷却至约15℃,将20v 5%tfa/dcm装入反应器,然后在氮气中在维持约15℃的温度下搅拌2小时。过滤树脂,并用3x10v dcm洗涤。将全部滤液合并在一起。在减压下将dcm从所得溶液中除去,同时保持内部温度≤20℃直至22.5v剩余
体积。将mtbe(25v)加入该溶液中,将dcm/mtbe溶剂再次减压除去,同时保持温度≤20℃直至22.5v剩余体积。重复加入mtbe/蒸馏操作,直至在上清液中的片段剩余浓度未达到《0.11重量%。然后,将所得浆体过滤,期间保持温度在约15℃。向该滤饼中加入14v新鲜的mtbe,并将该浆体在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得固体在约35℃下干燥。
[0226]
实施例11:中间体化合物11的固相肽合成
[0227]
中间体化合物11(seq id no:17)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-aib-2-ctc树脂(0.6-0.9mmol/g)进行,条件如下表11所示。
[0228]
表11:实施例11的spps条件
[0229][0230][0231]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中搅拌10-15分钟,保持温度在约25℃。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3v dcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤x%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0232]
实施例12:中间体化合物12的固相肽合成
[0233]
中间体化合物12(seq id no:18)或其可药用盐可通过标准spps合成。简言之,spps使用sieber树脂(负载系数0.6-0.9mmol/g)进行,条件如下表12所示。
[0234]
表12:实施例12的spps条件
[0235]
[0236]
[0237][0238]
fmoc脱保护、片段切割和分离:用10v 20%哌啶/dmf搅拌sieber树脂上的片段两次20-30分钟,然后用10v dmf洗涤六次。使用10v dcm将sieber树脂上的脱fmoc片段溶胀两次,持续10-20分钟。将装有树脂的反应器冷却至约15℃。将20v 5%tfa/dcm装入反应器,并在氮气中在约15℃的温度下搅拌2小时。过滤树脂,并用3x10v dcm洗涤。将全部滤液合并在一起。在减压下将dcm从所得溶液中除去,同时保持内部温度≤20℃直至22.5v剩余体积。将mtbe(25v)加入该溶液中,将dcm/mtbe溶剂再次减压除去,同时保持温度≤20℃直至22.5v剩余体积。重复加入mtbe/蒸馏操作,直至在上清液中的片段剩余浓度未达到《0.11重量%。然后,将所得浆体过滤,期间保持温度在约15℃。向该滤饼中加入14v新鲜的mtbe,并将该浆体在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得固体在约35℃下干燥。
[0239]
实施例13:中间体化合物13的固相肽合成
[0240]
中间体化合物13(seq id no:19)或其可药用盐可通过标准spps合成。简言之,spps使用sieber树脂(负载系数0.6-0.9mmol/g)进行,条件如表13所示。
[0241]
表13:实施例13的spps条件
[0242]
[0243]
[0244][0245]
fmoc脱保护、片段切割和分离:用10v 20%哌啶/dmf搅拌sieber树脂上的片段两次20-30分钟,然后用10v dmf洗涤六次。使用10v dcm将sieber树脂上的脱fmoc片段溶胀两次,持续10-20分钟。将装有树脂的反应器冷却至约15℃。将20v 5%tfa/dcm装入反应器,并在氮气中在约15℃的温度下搅拌2小时。过滤树脂,并用3x10v dcm洗涤。将全部滤液合并在一起。在减压下将dcm从所得溶液中除去,同时保持内部温度≤20℃直至22.5v剩余体积。将mtbe(25v)加入该溶液中,将dcm/mtbe溶剂再次减压除去,同时保持温度≤20℃直至22.5v剩余体积。重复加入mtbe/蒸馏操作,直至在上清液中的片段剩余浓度未达到《0.11重量%。然后,将所得浆体过滤,期间保持温度在约15℃。向该滤饼中加入14v新鲜的mtbe,并将该浆体在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得固体在约35℃下干燥。
[0246]
实施例14:中间体化合物14的固相肽合成
[0247]
中间体化合物14(seq id no:20)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-aib-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表14所示。
[0248]
表14:实施例14的spps条件
[0249]
[0250][0251]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3vdcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤x%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0252]
实施例15:中间体化合物15的固相肽合成
[0253]
中间体化合物15(seq id no:21)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-aib-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表15所示。
[0254]
表15:实施例15的spps条件
[0255]
[0256][0257]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3vdcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤x%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0258]
实施例16:中间体化合物的固相肽合成16
[0259]
中间体化合物16(seq id no:22)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-ala-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表16所示。
[0260]
表16:实施例16的spps条件
[0261]
[0262][0263]
片段切割和分离:将在ctc树脂上的片段使用dcm(5v)溶胀一次持续45分钟。将5v 1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中在约25℃下搅拌10-15分钟。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复两次。将该树脂用3vdcm洗涤,并搅拌10-15分钟。将全部滤液和洗涤液合并,并将所得混合物冷却至≤20℃。将该片段溶液真空浓缩至2-4v,保持温度≤20℃。向该溶液中加入5v acn,并真空除去剩余的dcm(剩余dcm浓度≤x%),保持温度≤20℃。将该片段的acn溶液历经2-6小时时间(《1l/min)加入5v冰冷却的h2o中,保持温度在约0℃。将所得沉淀片段的浆体在约0℃搅拌30-40分钟,然后在约0℃过滤。将所得固体在约25℃悬浮于3-5v h2o中,搅拌10-15分钟,然后过滤。再重复一次洗涤,并将所得固体在约40℃下干燥。
[0264]
实施例17:中间体化合物17的混合的液固相肽合成
[0265]
中间体化合物17(seq id no:23)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-aib-2-ctc-肼树脂(负载系数0.6-0.9mmol/g)进行,条件如下表17所示。
[0266]
表17:实施例17的spps条件
[0267]
[0268][0269]
使用过滤反应器用dcm(3x10v)溶胀树脂上的片段。通过混合10v tfa、0.4v tips、0.4v h2o和0.3重量v的dtt并搅拌至均匀以制备脱保护混合试剂。将该混合试剂加到树脂中,并在室温下将所得浆体搅拌3小时。过滤树脂并用dcm(2x3v)洗涤。然后,将所得滤液合并并冷却至约-10℃,缓慢加入75v mtbe。过滤所得浆体,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到白色固体产物。
[0270]
将粗的肽酰肼溶解在30v的连接缓冲液(6m盐酸胍和0.2m磷酸二氢钠缓冲液,ph 3.35)中,并冷却至约-15℃。向该酰肼溶液中加入1m亚硝酸钠溶液(5.0-10.0当量),并在约-15℃下搅拌10分钟。10分钟后,将2,2,2-三氟乙硫醇(20.0当量,ph 7.0)加入由肽酰肼氧化产生的肽基叠氮化物中。用5n氢氧化钠溶液将反应混合物的ph值调节至7.0。使该肽基叠氮化物的硫解进行1小时,然后将所得肽硫酯直接用于连接化学或通过反相色谱纯化(参见huang等人(2014)tetrahedron 70:2951-2955)。
[0271]
实施例18:中间体化合物18的固相肽合成
[0272]
中间体化合物18(seq id no:24)或其可药用盐可通过标准spps合成。简言之,
spps使用sieber树脂(负载系数0.6-0.9mmol/g)进行,条件如表18所示。
[0273]
表18:实施例18的spps条件
[0274]
0.9mmol/g)进行spps,条件如下表19所示。
[0279]
表19:实施例19的spps条件
[0280]
[0281][0282]
使用过滤反应器用dcm(3x10v)溶胀树脂上的片段。通过混合10v tfa、0.4v tips、0.4v h2o和0.3重量v的dtt并搅拌至均匀以制备脱保护混合试剂。将该混合试剂加到树脂中,并在室温下将所得浆体搅拌3小时。过滤树脂并用dcm(2x3v)洗涤。然后,将所得滤液合并并冷却至约-10℃,缓慢加入75v mtbe。过滤所得浆体,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到白色固体产物。
[0283]
将粗的肽酰肼溶解在30v的连接缓冲液(6m盐酸胍和0.2m磷酸二氢钠缓冲液,ph 3.35)中,并冷却至约-15℃。向该酰肼溶液中加入1m亚硝酸钠溶液(5.0-10.0当量),并在约-15℃下搅拌10分钟。10分钟后,将2,2,2-三氟乙硫醇(20.0当量,ph 7.0)加入由肽酰肼氧化产生的肽基叠氮化物中。用5n氢氧化钠溶液将反应混合物的ph值调节至7.0。使该肽基叠氮化物的硫解进行1小时,然后将所得肽硫酯直接用于连接化学或通过反相色谱纯化(参见huang(2014))。
[0284]
实施例20:中间体化合物20的固相肽合成
[0285]
中间体化合物20(seq id no:26)或其可药用盐可通过标准spps合成。简言之,spps使用sieber树脂(负载系数0.6-0.9mmol/g)进行,条件如表20所示。
[0286]
表20:实施例20的spps条件
[0287]
[0288][0289]
切割和脱保护:使用过滤反应器用dcm(3x10v)溶胀树脂上的片段。通过混合10v tfa、0.4v tips、0.4v h2o和0.3重量v的dtt并搅拌至均匀以制备脱保护混合试剂。将该混合试剂加到树脂中,并在室温下将所得浆体搅拌3小时。过滤树脂并用dcm(2x3v)洗涤。然后,将所得滤液合并并冷却至约-10℃,缓慢加入75v mtbe。过滤所得浆体,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到白色固体产物。
[0290]
实施例21:中间体化合物21的混合的液固相肽合成
[0291]
中间体化合物21(seq id no:27)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-lys(boc)-2-ctc-肼树脂(负载系数0.6-0.9mmol/g)进行,条件如下表21所示。
[0292]
表21:实施例21的spps条件
[0293]
[0294][0295]
使用过滤反应器用dcm(3x10v)溶胀树脂上的片段。通过混合10v tfa、0.4v tips、0.4v h2o和0.3重量v的dtt并搅拌至均匀以制备脱保护混合试剂。将该混合试剂加到树脂中,并在室温下将所得浆体搅拌3小时。过滤树脂并用dcm(2x3v)洗涤。然后,将所得滤液合并并冷却至约-10℃,缓慢加入75v mtbe。过滤所得浆体,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到白色固体产物。
[0296]
将粗的肽酰肼溶解在30v的连接缓冲液(6m盐酸胍和0.2m磷酸二氢钠缓冲液,ph 3.35)中,并冷却至约-15℃。向该酰肼溶液中加入1m亚硝酸钠溶液(5.0-10.0当量),并在约-15℃下搅拌10分钟。10分钟后,将2,2,2-三氟乙硫醇(20.0当量,ph 7.0)加入由肽酰肼氧化产生的肽基叠氮化物中。用5n氢氧化钠溶液将反应混合物的ph值调节至7.0。使该肽基叠氮化物的硫解进行1小时,然后将所得肽硫酯直接用于连接化学或通过反相色谱纯化(参见huang(2014))。
[0297]
实施例22:中间体化合物22的固相肽合成
[0298]
中间体化合物22(seq id no:28)或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-gly-2-ctc树脂(负载系数0.6-0.9mmol/g)进行,条件如下表22所示。
[0299]
表22:实施例22的spps条件
1%tfa/dcm装入反应器,并将所得树脂的混悬液在氮气中搅拌10-15分钟,期间保持温度在约25℃。移出滤液,并立即通过缓慢加入1.05当量吡啶进行中和。用1%tfa/dcm处理树脂、然后滤液中和,重复四次。将该树脂用3.5v dcm洗涤,并搅拌5-10分钟。将全部滤液和洗涤液合并。将片段溶液真空浓缩至1.5v,期间保持温度≤35℃。将5v ipac加入该溶液中,并真空除去剩余的ipac/dcm溶剂,期间保持温度≤40℃。重复加入5v ipac和真空蒸馏以产生3.5v的最终片段溶液。然后将该溶液用3x2v 5.0%nacl溶液洗涤,随后减压除去ipac至1.5v,期间保持温度≤40℃。在40℃下将4-5v庚烷加入该溶液中。然后,将温度降至约15℃,并将所得浆体搅拌30分钟。减压除去ipac/庚烷溶剂至3.5v,期间保持温度≤40℃。重复加入庚烷并蒸馏,将所得沉淀片段的浆体冷却至约20℃,过滤,用2v庚烷洗涤,并将所得固体在约35℃下干燥。
[0307]
实施例24:中间体化合物24的液相肽合成
[0308]
中间体化合物24或其可药用盐可使用标准偶联化学在溶液中通过h-l-2-me-leu和fmoc-l-ile-oh偶联合成,然后进行后处理和分离。
[0309]
实施例25:脂肪酸部分的固相肽合成(化合物25)
[0310]
化合物25
[0311]
或其可药用盐可通过标准spps合成。简言之,spps使用fmoc-peg-2-ctc树脂(负载系数0.6-1.1mmol/g)进行,条件如下表24所示。
[0312]
表24:实施例25的spps条件
[0313][0314]
实施例26:肠促胰岛素类似物从四种中间体化合物经化学偶联的混合的液固相合成
[0315]
偶联方案:seq id no:6的肠促胰岛素类似物可以经hlsps偶联seq id nos:7、8、9和10来制备。简言之,将seq id no:7(1.05-1.30mmol)的溶液与seq id no:8(1.00mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.30-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌2-4小时。然后,加入10当量dea,
并将该混合物搅拌4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0316]
接下来,将偶联的seq id nos:7 8(1.00mmol)溶液与seq id no:9(1.05-1.30mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.30-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌2-4小时。然后,加入10当量dea,并将该混合物搅拌2-4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0317]
接下来,将偶联的seq id nos:7 8 9(1.00mmol)溶液与seq id no:10(1.20-1.30mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.50-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌3-4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0318]
总体脱保护:通过将10v tfa、2v dcm、0.4v tips、0.4v h2o和0.3重量v dtt混合并搅拌直至均匀来制备脱保护混合试剂。将该混合试剂冷却至约15℃,然后加入固体的偶联的seq id nos:7 8 9 10,并将所得反应混合物加温至室温,并在室温下搅拌3小时。将该混合物冷却至约-10℃,并缓慢加入75v mtbe。将所得浆体过滤,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到产物seq id no:6,为白色固体。
[0319]
实施例27:肠促胰岛素类似物从四种中间体化合物经化学偶联的混合的液固相合成
[0320]
此处,基本上按照实施例26中所述用于seq id nos:7、8、9和10偶联的方法,seq id no:6的肠促胰岛素类似物通过片段缩合固相肽合成(cspps)将seq id nos:7、11、12和10偶联来制备。
[0321]
实施例28:肠促胰岛素类似物从四种中间体片段经化学偶联的混合的液固相合成
[0322]
此处,基本上按照实施例26中所述用于seq id nos:7、8、9和10偶联的方法,seq id no:6的肠促胰岛素类似物通过cspps将seq id nos:7、13、14和10偶联来制备。
[0323]
实施例29:肠促胰岛素类似物从三种中间体片段经化学偶联的混合的液固相合成
[0324]
seq id no:6的肠促胰岛素类似物可以经cspps偶联seq id nos:7、13和15来制备。简言之,将seq id no:7(1.05-1.30mmol)的溶液与seq id no:13(1.00mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.30-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌2-4小时。然后,加入10当量dea,并将该混合物搅拌4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0325]
接下来,将偶联的seq id nos:7 13(1.00mmol)溶液与seq id no:15(1.20-1.30mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.50-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌2-4小时。然后,加入10当量dea,并将该混合物搅拌2-4小时。将该混合物用20v 15-20%盐水溶液猝灭,
然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0326]
总体脱保护:通过将10v tfa、2v dcm、0.4v tips、0.4v h2o和0.3重量v dtt混合并搅拌直至均匀来制备脱保护混合试剂。将该混合试剂冷却至约15℃,然后加入固体的偶联的seq id nos:7 13 15,并将所得反应混合物加温至室温,并在室温下搅拌3小时。将该混合物冷却至约-10℃,并缓慢加入75v mtbe。将所得浆体过滤,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到产物seq id no:6,为白色固体。
[0327]
实施例30:肠促胰岛素类似物从三种中间体片段经化学偶联的混合的液固相合成
[0328]
seq id no:6的肠促胰岛素类似物可以经cspps偶联seq id nos:16、9和10来制备。简言之,将seq id no:16(1.00mmol)的溶液与seq id no:9(1.05-1.30mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.30-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌3-4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0329]
接下来,将偶联的seq id nos:16 9(1.00mmol)溶液与seq id no:10(1.20-1.30mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.50-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌2-4小时。然后加入10当量dea,并将该混合物搅拌2-4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0330]
总体脱保护:通过将10v tfa、2v dcm、0.4v tips、0.4v h2o和0.3重量v dtt混合并搅拌直至均匀来制备脱保护混合试剂。将该混合试剂冷却至约15℃,然后加入固体的偶联的seq id nos:16 9 10,并将所得反应混合物加温至室温,并在室温下搅拌3小时。将该混合物冷却至约-10℃,并缓慢加入75v mtbe。将所得浆体过滤,并用2x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到产物seq id no:6,为白色固体。
[0331]
实施例31:肠促胰岛素类似物从三种中间体片段经化学偶联的混合的液固相合成
[0332]
此处,基本上按照实施例30中所述用于seq id nos:16、9和10偶联的方法,seq id no:6的肠促胰岛素类似物通过cspps将seq id nos:18、12和10偶联来制备。
[0333]
实施例32:肠促胰岛素类似物从两种中间体片段经化学偶联的混合的液固相合成
[0334]
seq id no:6的肠促胰岛素类似物可以经cspps偶联seq id nos:19和15来制备。简言之,将seq id no:18(1.00mmol)的溶液与seq id no:15(1.20-1.30mmol)的溶液在30-40v dmso/acn(70:30)中使用pybop、hatu或pyoxim试剂(1.50-2.00mmol)和diea(4.00-5.00mmol)在室温下偶联。将该混合物在室温下搅拌2-4小时。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外的10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将固体在真空干燥器(40℃)中干燥,得到产物,为白色固体。
[0335]
总体脱保护:通过将10v tfa、2v dcm、0.4v tips、0.4v h2o和0.3重量v dtt混合并搅拌直至均匀来制备脱保护混合试剂。将该混合试剂冷却至约15℃,然后加入固体的偶联的seq id nos:19 15,并将所得反应混合物加温至室温,并在室温下搅拌3小时。将该混合物冷却至约-10℃,并缓慢加入75v mtbe。将所得浆体过滤,并用2x10v mtbe洗涤。将该固
体在真空干燥器(40℃)中干燥,得到产物seq id no:6,为白色固体。
[0336]
实施例33:肠促胰岛素类似物从两种中间体片段经化学偶联的混合的液固相合成
[0337]
此处,基本上按照实施例32中所述用于seq id nos:15和19偶联的方法,seq id no:6的肠促胰岛素类似物通过cspps将seq id nos:18和20偶联来制备。
[0338]
实施例34:肠促胰岛素类似物从两种中间体片段经化学偶联的混合的液固相合成
[0339]
此处,基本上按照实施例32中所述用于seq id nos:15和19偶联的方法,seq id no:6的肠促胰岛素类似物通过cspps将seq id nos:21和18或seq id nos:22和19偶联来制备,唯一不同是在两个片段偶联后,通过化学转化(条件取决于基团的性质)选择性地去除lys17上的保护基团,并随后与脂肪酸侧链选择性酰化,然后总体脱保护。
[0340]
实施例35:肠促胰岛素类似物从两种中间体片段经天然化学连接的混合的液固相合成
[0341]
seq id no:6的肠促胰岛素类似物可以经天然化学连接偶联seq id nos:23和24来制备。简言之,将seq id nos:23的肽硫酯溶于30-50v连接缓冲液(6m盐酸胍和0.2m磷酸二氢钠缓冲液,ph 7.04)。将含有n-末端半胱氨酸的肽片段seq id nos:24(0.9-0.95当量)加入所述硫酯溶液中。向反应混合物中加入40当量的2,2,2-三氟乙硫醇(ph 7.16)和20当量的三(2-羧基乙基)膦(ph7.0),并用5n氢氧化钠溶液将ph调至7.0。将反应物在室温下搅拌24小时,所得溶液直接用于反相纯化。
[0342]
实施例36:肠促胰岛素类似物从两种中间体片段经天然化学连接的混合的液固相合成
[0343]
此处,基本上按照实施例35中所述用于seq id nos:23和24偶联的方法,seq id no:6的肠促胰岛素类似物通过cspps将seq id nos:25和26偶联来制备。
[0344]
实施例36:化合物35的合成
[0345][0346]
15-30℃下将二氯甲烷(7.5l,15.0体积)加入20l四颈瓶中,并在15-30℃下加入18-(叔丁氧基)-18-氧代十八烷酸(500.5g,1.0当量1.35mol)和n-羟基琥珀酰亚胺(185.6g,1.2当量,1.61mol),得到混悬液。将反应混合物冷却至0-10℃,并一次加入n-乙基-n
′‑
碳二亚胺(338.4g,1.3当量,1.77mol),得到溶液。用8体积的半饱和盐水洗涤反应混合物三次。化合物35直接用于下一步骤以制备化合物36。
[0347]
化合物35的替代方法
[0348]
将18-(叔丁氧基)-18-氧代十八烷酸(20g,53.431mmol,99质量%)、n,n'-二琥珀酰亚胺基碳酸酯(1.2当量,64.117mmol,99.6质量%)和4-二甲基氨基吡啶(0.2当量,1.31g,10.7mmol,100质量%)装入配有顶置式搅拌器的1000ml带挡板的夹套反应器中。加入乙酸乙酯(800ml,40体积),并将所得浆体在环境温度(18℃
–
23℃)下搅拌过夜(18-24h)。24h后粗样品的1h-nmr通常显示97-99%反应完成。用去离子水(3x82ml)萃取该批次。将有机层真空浓缩至~160ml。向反应物中加入乙酸乙酯(200ml),并在50℃下真空中将粗反应
溶液减少至~180ml。将溶液转移至夹套过滤器中,并在搅拌的同时缓慢冷却至3℃。然后将反应物在3-5℃保持1h。过滤固体,用冷的乙酸乙酯(18ml)洗涤,并真空干燥(7英寸汞柱),得到固体的化合物35(22.6g,90.5%产率,99.17%hplc-cad)。
[0349]
实施例37:合成t-buo-c18-glu-1-otbu(化合物36)
[0350][0351]
15-30℃下在20l四颈瓶中将化合物35加入(4s)-4-氨基-5-叔丁氧基-5-氧代-戊酸(h-glu-1-otbu)(289g,1.14当量,1mol)在二氯甲烷(2.5l,5.0体积)的溶液中。然后在15-30℃下在反应器中加入二异丙基乙胺(230g,1.5当量,1.78mol),得到溶液。一旦制备1《0.5%,反应继续进行下一步骤。用2%的khso4水溶液(4g/g x 3)洗涤有机相,并在真空下在t《50℃和《-0.08mpa下浓缩至1-2体积。向反应器中加入乙腈(8体积),并在《60℃下浓缩至1-2体积。将乙腈(8体积)再次加入反应器并在《60℃下浓缩至5-6体积。将浓缩物冷却至40-50℃,搅拌0.5-1小时,然后冷却至15-30℃,持续2-4小时。过滤该浆体,用乙腈(3体积)洗涤,并在n2下干燥,得到固体的化合物36(659.5g,86.3%产率,99.1%lcap)。
[0352]
化合物36的替代方法
[0353]
将化合物35(50g,104.8mmol,98质量%)、(4s)-4-氨基-5-叔丁氧基-5-氧代-戊酸h-glu-1-otbu(h-glu-1-otbu)(25.7g,126mmol,99.3质量%)装入配有顶置式搅拌器和热电偶的1l带挡板的夹套反应器中。使用乙腈(500ml,10v)将固体沿着漏斗冲洗至反应容器中。然后将二异丙基乙胺(22ml,126mmol,99.75质量%)加入到反应物中。将反应加热至40℃并搅拌18小时。通过1h-nmr/hplc-cad确认反应完成后,将乙酸(7.2ml,130mmol,100质量%)和水(215ml,11934.6mmol,100质量%)加入反应物中,并在30-35℃下搅拌1小时。将该批次转移至配有顶置式搅拌器的夹套过滤器中,并冷却至-20℃。在tr=2℃和tj=-9℃时开始从溶液中结晶出固体。将该固体在此温度下保持1小时。然后将去离子水(8.6v,460ml)倒入该批次中,将过滤器加温至0℃。将固体过滤并在40℃在高真空下干燥,获得固体的化合物36(55.5g,95.3%产率,99.62%hplc-cad)。
[0354]
实施例38:合成t-buo-c18-glu-1-otbu-5-onsu(化合物37)
[0355][0356]
在15-30℃下将乙腈(12.0体积)加入20l四颈瓶中。在15-30℃下将化合物36(500.4g,1.0当量,0.90mol)和n,n'-二琥珀酰亚胺基碳酸酯(278.5g,1.2当量,1.09mol)加
入该烧瓶中,得到混悬液。将4-二甲基氨基吡啶(11.0g 0.1当量0.09mol)一次性加入,得到溶液。历经0.5-1小时加入水(1.6kg)。将该混合物冷却至0-10℃持续1-2小时,过滤,用乙腈(2体积0-10℃)洗涤,并在n2下干燥,得到固体的化合物37(536.0g,91.3%产率100.0%lcap)。
[0357]
化合物37的替代合成
[0358]
将化合物36(55g,98.96mmol)、n,n'-二琥珀酰亚胺基碳酸酯(31g,121mmol,99.6质量%)、4-二甲基氨基吡啶(1.22g,9.89mmol,99质量%)装入配有顶置式搅拌器和热电偶的1l带挡板的夹套反应器中。将乙腈(660ml,12v)加入该反应容器中。将反应物在24℃搅拌4小时。通过1h-nmr/hplc-cad确认反应完成后,将反应溶液转移至1000ml烧杯中,并将去离子水(180ml)加至该配有磁力搅拌棒的烧杯中。溶液搅拌时固体从反应物中迅速产生。将反应物浆体在冰箱(2-10℃)中冷却过夜。过滤固体,并将滤饼用125ml冷却的(2-10℃)乙腈洗涤。将固体在40℃于高真空下干燥24小时形成化合物37(59.3g,91.8%产率,99.61%hplc-cad)。
[0359]
实施例39:合成t-buo-c18-glu-1-otbu-5-(aeea)2(化合物38)
[0360][0361]
15-30℃下将二氯甲烷(7.5l,15.0体积)一次性加入20l四颈瓶中,然后在15-30℃下加入(aeea)2(261g,1.1当量,0.85mol)、化合物37(501g,1.0当量,0.77mol)和二异丙基乙胺(1.5当量)。一旦化合物37《0.5%,反应继续下一步骤。然后反应混合物在t《30℃、p《-0.08mpa下于真空中浓缩至5-6体积。将乙酸乙酯加入该粗产物(5体积),在t《50℃、p《-0.08mpa下真空浓缩。将乙酸乙酯(10体积)加入该浓缩物,并用2%khso4水溶液(5g/g x 5-6)洗涤,在t《40℃和p《-0.08mpa下于真空中浓缩至1.2体积。将二甲基甲酰胺加入该浓缩物(3g/g体积),得到产物化合物38,为浅黄色溶液(2.4kg,92.7%产率,98.7%lcap)。
[0362]
化合物38的替代合成
[0363]
将(aeea)2(27.6g,1.1当量,85.0mmol,95质量%)、n-甲基-n-三甲基硅烷基乙酰胺(30ml,2当量,200mmol,90质量%)和乙酸乙酯(230ml)加入配有热电偶和磁力搅拌棒的500ml烧瓶中。将反应物在18-23℃搅拌3小时。3小时后,将化合物37(50g,76.58mmol,100质量%)和乙酸乙酯(130ml)加入该反应瓶中,并在18-23℃搅拌2小时。通过1h-nmr/hplc-cad确认反应完成后,将反应溶液转移至分液漏斗,并将有机层用2%khso4溶液(100ml x 3)和2%nacl溶液(100ml x 6)洗涤。将有机层浓缩,并将所得油状物在50℃高真空浓缩24小时,得到化合物38,在-20℃为蜡状固体(66.32g,94.9%q-nmr效价,99.53%hplc-cad)。
[0364]
实施例40:天然化学连接
[0365][0366]
化合物39合成
[0367]
将fmoc-肼-2-氯三苯甲基树脂(1.16g,0.85mmol)在symphony x合成器上用2x10ml dmf溶胀每次20分钟。每次用3x10ml 20%哌啶/dmf进行fmoc脱保护30分钟。然后用5x10ml dmf洗涤该树脂。
[0368]
将(2s)-6-[[2-[2-[2-[[2-[2-[2-[[(4s)-5-叔丁氧基-4-[(18-叔丁氧基-18-氧代-十八酰基)氨基]-5-氧代-戊酰基]氨基]乙氧基]乙氧基]乙酰基]氨基]乙氧基]乙氧基]乙酰基]氨基]-2-(9h-芴-9-基甲氧基羰基氨基)己酸(化合物38,2.13g,1.78mmol,2.1当量)和tntu(0.715g,1.96mmol,2.31当量)溶于约20.5ml dmf中,并将n,n-二异丙基乙胺(0.57ml,3.27mmol,3.85当量)加入该溶液中。将该溶液在旋转混合器上混合15分钟。15分钟后,将该预活化的化合物38的溶液加入树脂中,并将偶联进行8小时。然后,将树脂用5x10ml dmf、5x10ml dcm洗涤,并在合成器上干燥8小时。树脂负载通过定量nmr测定为0.45mmol/g。
[0369]
实施例41:化合物40(seq id no:58)合成
[0370]
将每个约1.10g化合物39(负载值:0.45mmol/g)加入两个40ml反应容器中,用2x20ml dmf溶胀每次20分钟。使用标准spps方案合成seq id no:58。
[0371]
脱保护:4x9ml 20%v/v哌啶在dmf中的溶液,各30分钟。
[0372]
偶联:3当量氨基酸、3当量oxyma和3.3当量dic用于氨基酸偶联。
[0373]
在spps过程中,在每次偶联和最终重复fmoc脱保护后,都用5x9ml dmf洗涤树脂、同时用1min n2混合。在肽酰肼合成结束时,用dcm洗涤树脂、同时用n2混合。将树脂在肽合成器上干燥。
[0374]
总体脱保护和切割
[0375]
将45ml由2.5%w/v二硫苏糖醇(dtt)、2.5%v/v水、2.5%v/v三异丙基硅烷(tips)
和92.5%三氟乙酸(tfa)制备的切割试剂混合物加入在500ml三颈圆底烧瓶中的干燥树脂(4.2g)中,并搅拌约3小时。将树脂过滤,并用2x2.5ml tfa洗涤。将滤液倒入350ml冷却的mtbe中,肽立即沉淀析出。用2x2.5ml tfa洗涤该过滤瓶,并倒入所述冷却的mtbe中。将其冷却至-20℃半小时,然后离心。然后用300ml mtbe洗涤该肽沉淀物两次并离心。将该肽沉淀物在27℃在真空烘箱中干燥约14小时。干燥后得到大约2.75g粗的化合物40[预期值(质量 2h
)/2=1356.2257,实测值(质量 2h
)/2=1356.2245]。
[0376]
实施例42:化合物41(seq id no:59)的合成
[0377]
与化合物40,seq id no:58的合成类似,通过标准spps方案在sieber酰胺树脂上合成约0.50mmol化合物41(seq id no:59)。
[0378]
总体脱保护和切割:将25ml用5%w/v二硫苏糖醇(dtt)、2.5%v/v水、2.5%v/v三异丙基硅烷(tips)和90%三氟乙酸(tfa)制备的切割试剂混合物加入干燥树脂(2.21g)中,在旋转混合器上混合3小时。将树脂过滤,并用2x2.0ml tfa洗涤。将滤液倒入175ml冷却的mtbe中,肽立即沉淀析出。用2x2ml tfa洗涤该过滤瓶,并倒入所述冷却的mtbe中。将其冷却至-20℃ 30分钟,然后离心。然后用150ml mtbe洗涤该肽沉淀物两次并离心。该肽沉淀物在27℃在真空烘箱中干燥约14小时。干燥后得到约1.351g粗的化合物41(seq id no:58)。在环境温度下,通过rp-hplc在waters csh c18 10μm柱(10mm x250mm)上纯化,使用线性梯度:最初3分钟为10%乙腈的水溶液(0.1%tfa)、然后10-15%乙腈的水溶液(0.1%tfa)3-5分钟、随后15-35%乙腈的水溶液(0.1%tfa)23分钟。得到约650mg纯化的化合物41[预期值(质量 2h
)/2=1138.5486,实测值(质量 2h
)/2=1138.5458]。
[0379]
实施例43:化合物42(seq id no:60)的合成
[0380]
化合物42(硫酯合成),seq id no:60
[0381]
将粗的肽酰肼(化合物40;seq id no:58,118.2mg,0.044mmol)溶解于10ml连接缓冲液(6m盐酸胍和0.3m磷酸二氢钠,ph约3.5)中。将该溶液在丙酮冰浴中冷却至
–
15℃。将0.3ml 1m亚硝酸钠溶液(20.7mg,0.3mmol,6.8当量)加入到肽酰肼溶液中,并在-15℃下搅拌15分钟。同时,用连接缓冲液(ph约7.0)将0.2ml硫酚稀释至1.1ml。15分钟后,向肽酰肼溶液中加入1.1ml的硫酚混合物,以使得化合物40生成的肽基叠氮化物在原位硫解。
[0382]
用5n氢氧化钠溶液将反应混合物的ph值调至约7.0。使肽基叠氮化物硫解15分钟,得到化合物42(seq id no:60)。
[0383]
实施例44:化合物43,seq id no:61(与硫酯化合物42的天然化学连接):
[0384]
将化合物41(seq id no:59(75.4mg,0.033mmol)在闪烁瓶中溶于2ml连接缓冲液(ph约7.0),并将该溶液加入粗的硫酯化合物42溶液(seq id no:60)中。将小瓶用1ml连接缓冲液(ph约7.0)冲洗,并将冲洗液加入反应混合物中。将1.5ml三(2-羧基乙基)膦(tcep,0.25m在连接缓冲液中,ph约7.0)和1.0ml抗坏血酸溶液(0.53m在连接缓冲液中,ph约7.0)加入反应混合物中。用5nnaoh溶液将反应混合物的ph调至约7.1,该溶液变澄清。9-10小时内反应完成,得到seq id no:61。
[0385]
实施例45:seq id no:62(化合物44)的合成
[0386]
表25:seq id no:62(化合物44)的spps条件
[0387][0388]
片段切割和分离:ctc树脂上的片段用dcm溶胀两次。将装有树脂的反应器冷却至约15℃,并将2%tfa/dcm(4ml/g树脂)加入反应器,然后在氮气下搅拌15分钟。然后加入1%tfa/dcm(4ml/g树脂),搅拌15分钟,过滤后再重复。过滤树脂,用3x10v dcm洗涤。将所有滤液合并在一起,用dipea中和。从所得溶液中除去dcm,并加入盐水以沉淀片段2。然后,在将温度保持在约15℃下过滤所得浆体。向滤饼中加入14v新鲜mtbe,在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得淡黄色固体在约35℃下干燥。
[0389]
实施例46:seq id no:42(化合物45)合成
[0390]
表26:seq id no:42(化合物45)合成的spps条件
[0391][0392][0393]
片段切割和分离:ctc树脂上的片段用dcm溶胀两次。将装有树脂的反应器冷却至约15℃,并将20%hfip/dcm(8ml/g树脂)加入反应器,然后在氮气下搅拌60分钟。然后加入20%hfip/dcm(4ml/g树脂),搅拌60分钟。过滤树脂,用3x10v dcm洗涤。将所有滤液合并在一起,并从所得溶液中除去dcm和hfip,将庚烷和乙醚加入以沉淀片段3。然后,在将温度保持在约15℃下过滤所得浆体。向滤饼中加入14v新鲜mtbe,在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得橙色固体在约35℃下干燥。
[0394]
实施例47:seq id no:31(化合物28)合成
[0395]
表27:seq id no:31合成的spps条件
[0396][0397][0398]
片段切割和分离:ctc树脂上的片段用dcm溶胀两次。将装有树脂的反应器冷却至约15℃,并将2%tfa/dcm(4ml/g树脂)加入反应器,然后在氮气下搅拌15分钟。然后加入1%tfa/dcm(4ml/g树脂),搅拌15分钟,过滤后再重复。过滤树脂,用3x10v dcm洗涤。将所有滤
液合并在一起,用dipea中和。从所得溶液中除去dcm,并加入盐水以沉淀片段2。然后,在将温度保持在约15℃下过滤所得浆体。向滤饼中加入14v新鲜mtbe,在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得灰白色固体在约35℃下干燥。
[0399]
实施例48:seq id no:43(化合物46)的合成
[0400]
表28:经spps制备seq id no:43
[0401]
[0402][0403]
[0404]
片段切割和分离:用10v dcm溶胀sieber树脂上的片段两次10-20分钟。将装有树脂的反应器冷却至约15℃,并用6%tfa/dcm(5ml/g树脂)15min、3%tfa/dcm(5ml/g树脂)15min、1%tfa/dcm(10ml/g树脂)5min、1%tfa/dcm(5ml/g树脂)3min和1%tfa/dcm(2.5ml/g树脂)3min依次洗涤。过滤树脂,并用3x10v dcm洗涤。将全部滤液合并在一起。在减压下将dcm从所得溶液中除去,并用etoac重配。将庚烷加入该溶液,将所得浆体过滤,期间保持温度在约15℃。向该滤饼中加入14v新鲜的mtbe,在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得灰白色固体在约35℃下干燥。
[0405]
实施例49:seq id no:44(化合物47)的合成
[0406]
表29:seq id no:44制备
[0407]
[0408][0409]
片段切割和分离:用10v dcm溶胀sieber树脂上的片段两次10-20分钟。将装有树脂的反应器冷却至约15℃,并用6%tfa/dcm(5ml/g树脂)15min、3%tfa/dcm(5ml/g树脂)15min、1%tfa/dcm(10ml/g树脂)5min、1%tfa/dcm(5ml/g树脂)3min和1%tfa/dcm(2.5ml/g树脂)3min依次洗涤。过滤树脂,并用3x10v dcm洗涤。将全部滤液合并在一起。在减压下将dcm从所得溶液中除去,并用etoac重配。将庚烷加入该溶液,将所得浆体过滤,期间保持温度在约15℃。向该滤饼中加入14v新鲜的mtbe,在约15℃搅拌30分钟,然后过滤。再重复洗涤一次,并将所得灰白色固体在约35℃下干燥。
[0410]
实施例50:制备seq id no:29
[0411]
从四种中间体片段通过化学偶联的seq id no:29的混合的液固相合成
[0412]
seq id no:29可经hlsps由seq id nos:7、42、31和62偶联来制备。seq id no:7和62偶联生成seq id no:43。seq id no:42和31偶联生成seq id no:44。seq id no:43和seq id no:44偶联生成seq id no:29。
[0413]
简言之,将seq id no:7(1.00mmol)溶液和seq id no:62(1.1mmol)溶液在10v dmso中使用pybop、depbt或edc/honb试剂(1.30-2.00mmol)和diea(2mmol)在室温下偶联。将该混合物在室温下搅拌直至经hplc确认反应完成。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将该固体在真空干燥器(40℃)中干燥,得到seq id no:43,为灰白色固体。
[0414]
接下来,将seq id no:31(1.00mmol)溶液和seq id no:42(1.1mmol)溶液在x v dmso中使用pybop、depbt或edc/honb试剂(1.30-2.00mmol)和diea(2mmol)在室温下偶联。将该混合物在室温下搅拌直至经hplc确认反应完成。将该混合物用20v 15-20%盐水溶液猝灭,然后加入额外10v水,并搅拌10分钟。将所得浆体过滤,并将固体用3x10v水洗涤。将该固体在真空干燥器(40℃)中干燥,得到seq id no:44,为灰白色固体。
[0415]
最后,将seq id no:43(1.00mmol)溶液和seq id no:44(1.0mmol)溶液在17v thf中使用depbt试剂(1.30-2.00mmol)和diea(1mmol)在室温下偶联。将该混合物在室温下搅拌直至经hplc确认反应完成。将混合物用20v水猝灭。将所得浆体过滤,将固体用3x10v水洗涤。将该固体在真空干燥器(40℃)中干燥,得到白色固体产物。将所得肽用每克原料对应10ml tfa:tips:dtt:水(92.5:2.5:2.5:2.5)混合物进行脱保护。室温下搅拌3小时后,将产物用每ml混合物对应4ml 20%庚烷/mtbe沉淀,期间保持温度低于30℃。将所得浆体过滤,并将固体用3x10v mtbe洗涤。将该固体在真空干燥器(40℃)中干燥,得到产物,为灰白色固体。
[0416]
实施例51:seq id no:7的替代合成
[0417]
表30:该合成使用fmoc-sieber酰胺树脂,具有0.80mmol/g负载量。使用通常的spps方法,并具有以下修改:
[0418]
[0419][0420]
[0421]
将结合seq id no:7的树脂(54g,~24.0mmol)用2x300ml(各30min)20%pip/dmf处理。2)用6x300ml dmf,然后用5x300ml dcm洗涤。3)加入500ml tfa/dcm(5/95,v/v)并搅拌2小时。4)过滤该混合物,并用500ml dcm洗涤,得到1000ml体积的总滤液。5)浓缩至~250ml。6)加入250ml mtbe。7)重复步骤5-6共5-6次。8)过滤并收集湿滤饼,并在真空烘箱中于33℃干燥过夜,以产生seq id no:7(18.3g,75%产率),为白色固体。用uplc分析所分离的固体(94.4面积%)。lc-ms([m h]
):1020.58。
[0422]
实施例52:seq id no:45(化合物48)合成
[0423]
表31:该合成使用2-ctc树脂,具有0.80mmol/g负载量。使用通常的spps方法,并具有以下修改:
[0424]
[0425][0426][0427]
seq id no:45软切割:
[0428]
1)加入seq id no:45结合的树脂(4.0g,~1.34mmol)并加入40ml切割试剂混合物tfa/dcm(1/99,v/v/v)。2)室温下将其搅拌10分钟。3)过滤并收集滤液。4)用0.44ml吡啶(1/1,mol/mol)中和该滤液。5)再重复步骤1-4共3次。6)浓缩该合并滤液至干。7)用10ml dmso溶解该浆体。8)将该dmso溶液在搅拌下缓慢加入冷却的100ml水中。9)过滤并收集沉淀。10)用50ml水再重复浆化2次。11)真空干燥过夜,产生白色固体的seq id no:45(2.5g,58%产率)。使用uplc分析所分离的固体(95.0面积%)。lc-ms([m 2h]2
/2):1604.97。
[0429]
实施例53:seq id no:10的替代合成
[0430]
表32:该合成使用fmoc-leu-oh 2-ctc树脂,具有0.80mmol/g负载量。使用通常的spps方法,并具有以下修改:
[0431]
[0432][0433]
软切割:1)加入seq id no:10结合的树脂(60g,~40mmol)并加入600ml切割试剂混合物tfa/dcm(1/99,v/v/v)。2)室温下将其搅拌10分钟。3)过滤并收集滤液。4)用6.6ml吡
啶(1/1,mol/mol)中和该滤液。5)再重复步骤1-4共3次。6)浓缩该合并滤液至干。7)用60ml dmso溶解该浆体。8)将该dmso溶液在搅拌下缓慢加入冷却的600ml水中。9)过滤并收集沉淀。10)用300ml水再重复浆化2次。11)真空干燥过夜,产生湿固体的seq id no:10(56.4g,125%产率)。使用uplc分析所分离的固体(93.1面积%)。lc-ms([m h]
):2341.52。
[0434]
实施例54:经lpps合成seq id no:46(化合物49):
[0435]
向20ml玻璃闪烁瓶中加入seq id no:7(250mg,77.9μmol)、seq id no:45(103mg,90.7μmol)和dmso(5ml)。将diea(81μl,0.47mmol)加入该溶液中,然后加入pyaop(7-氮杂苯并三唑-1-基氧基)三吡咯烷子基磷鎓六氟磷酸盐(100mg,192μmol)。搅拌该反应物4小时,然后缓慢加入35ml冷水。经过滤收集沉淀的产物,随后用水(2x35ml)洗涤。真空干燥该湿滤饼,得到seq id no:46(化合物49),为白色固体(199mg,61%产率)。uplc:46.2面积%。
[0436]
实施例55:经lpps合成seq id no:47(化合物50):
[0437]
向20ml玻璃闪烁瓶中加入seq id no:46(500mg,120μmol),然后加入2ml mecn(2ml)。加入et2nh(0.5ml,4.8mmol),并搅拌4小时。浓缩该溶液至干。加入5ml mecn,再次浓缩至干。重复加入mecn和干燥2-3次。加入1ml mecn以溶解该浆体,然后将反应溶液在搅拌中缓慢加入15ml冷却的mtbe中。过滤并收集沉淀,并用10ml mtbe重新浆化2次。真空干燥该湿滤饼,得到seq id no:47(化合物50),为白色固体(200mg,42%产率)。uplc:75.3面积%。lc-ms[m 3h]
3
/3:1331.60。
[0438]
实施例56:经lpps合成seq id no:48(化合物51):
[0439]
向20ml玻璃闪烁瓶中加入seq id no:10(75mg,32.1μmol)、seq id no:47(100mg,25.0μmol)、1-羟基-7-氮杂苯并三唑(hoat;5mg,36.8μmol)和dmso(2ml)。将diea(30μl,173μmol)加入该溶液中,然后加入pyaop(33mg,63μmol)。搅拌该反应物5小时,然后缓慢加入15ml冷水。经过滤收集沉淀的产物,并随后用水(3x10ml)洗涤。真空干燥该湿滤饼,得到seq id no:48(化合物51),为白色固体(120mg,76%产率)。uplc:53.6面积%。
[0440]
实施例57:通过总体脱保护合成seq id no:6
[0441]
加入1ml切割试剂混合物溶液tfa/h2o/tips/dtt(0.925/0.025/0.025/0.025,v/v/v/v),然后将seq id no:48的样品(76mg,12.0μmol)加入该混合物中以得到溶液。将该混合物在约环境温度下搅拌约3小时。将该反应混合物倒入-15℃ mtbe(10ml)中,搅拌所得混悬液约30分钟。经过滤器过滤,并用mtbe(2x10 ml)洗涤该湿滤饼。将湿滤饼在35℃真空干燥,得到seq id no:6(80mg,44.8面积%,140%粗产率),为湿的固体。uplc:44.8面积%。lc-ms[m 3h]
3
/3:1183.20。
[0442]
实施例58:seq id no:11的替代合成
[0443]
表33:该合成使用fmoc-gly-ctc树脂,具有0.835mmol/g负载量。使用通常的spps方法,并具有以下修改。
[0444][0445][0446]
软切割:1)加入seq id no:11结合的树脂(10.0g,~4.4mmol)并加入100ml切割试剂混合物tfa/dcm(1/99,v/v/v)。2)室温下将其搅拌10分钟。3)过滤并收集滤液。4)用1.1ml吡啶(1/1,mol/mol)中和该滤液。5)再重复步骤1-4共3次。6)浓缩该合并滤液至干。7)用20ml dmso溶解该浆体。8)将该dmso溶液在搅拌下缓慢加入冷却的100ml水中。9)过滤并收集沉淀。10)用100ml水再重复浆化2次。11)真空干燥过夜,产生白色固体的seq id no:11(4.01g,62%产率)。使用uplc分析所分离的固体(97.6面积%)。lc-ms[m h]
:1445.78。
[0447]
实施例59:seq id no:18的替代合成
[0448]
表34:该合成使用fmoc-sieber酰胺树脂,具有0.71mmol/g负载量。使用通常的spps方法,并具有以下修改:
[0449]
[0450]
[0451][0452]
软切割:1)在最后的2x30min脱fmoc循环后,将结合seq id no:18(8.2g,~3.1mmol)的树脂加入40ml切割试剂混合物tfa/hfip/dcm(1/25/74,v/v/v)中,并将其在25℃搅拌5分钟。2)过滤并收集滤液,用0.44ml吡啶(1/1,mol/mol)中和该滤液。3)再重复该切割过程2次。4)浓缩该合并滤液至干。5)用10ml dmso溶解该浆体,并将该dmso溶液在搅拌下
缓慢加入200ml mtbe中。6)过滤并收集沉淀。7)用40ml mtbe再重复浆化2次,过滤沉淀。8)真空干燥该粗物质过夜,得到2.76克粗物质(41.4%产率),hplc检测纯度为92.5%。lc-ms[m 2h]
2
/2:1113.90。
[0453]
实施例60:seq id no:20的替代合成
[0454]
表35:该合成使用2-ctc树脂,具有0.80mmol/g负载量。使用通常的spps方法,并具有以下修改。
[0455]
[0456]
[0457][0458]
软切割:1)加入seq id no:20结合的树脂(5.7g,~10mmol)并加入60ml切割试剂混合物tfa/dcm(1/99,v/v/v)。2)室温下将其搅拌10分钟。3)过滤并收集滤液。4)用6.6ml吡啶(1/1,mol/mol)中和该滤液。5)再重复步骤1-4共3次。6)浓缩该合并滤液至干。7)用30ml dmso溶解该浆体。8)将该dmso溶液在搅拌下缓慢加入冷却的300ml水中。9)过滤并收集沉淀。10)用200ml水再重复浆化2次。11)真空干燥过夜,产生白色固体的seq id no:20(4.5g,63.4%产率)。使用uplc分析所分离的固体(99.4面积%)。lc-ms[m 2h]
2
/2:2053.39。
[0459]
实施例61:经lpps合成seq id no:49(化合物52):
[0460]
向20ml玻璃闪烁瓶中加入seq id no:11(1.0当量,145mg)、seq id no:7(1.1当量,125mg)和dmso/mecn(5ml,4/1,v/v)以溶解全部物质。将diea(3.0当量,0.055ml)加入该反应混合物中,然后加入pyoxim(1.5当量,80mg)。搅拌该反应物4小时,然后在搅拌下缓慢加入40ml水。经过滤收集沉淀的产物,并随后用水(2x40ml)洗涤。真空干燥该湿滤饼,得到粗的固体seq id no:49,为白色固体(180mg,73.2%产率),hplc检测纯度为89.5%。lc-ms[m 2h]
2
/2:1225.2。
[0461]
实施例62:经lpps合成seq id no:18:
[0462]
向20ml玻璃闪烁瓶中加入seq id no:49(700mg),然后加入8ml dmso(2ml)。加入et2nh(2.0ml),并搅拌4小时。浓缩该溶液至干。在搅拌下加入60ml冷却的mtbe。过滤并收集沉淀,并用60ml mtbe重新浆化2次。真空干燥该湿滤饼,得到seq id no:18,为白色固体(520mg,82%产率),hplc检测纯度为82.3%。lc-ms[m 2h]
2
/2:1113.8。
[0463]
实施例63:经lpps合成seq id no:48:
[0464]
向20ml玻璃闪烁瓶中加入seq id no:20(1.0当量,84mg)、seq id no:18(1.1当量,50mg)、1-羟基-7-氮杂苯并三唑(hoat;1.0当量,3mg)和dmso(2ml)以溶解全部物质。将
diea(6当量,21μl)加入该溶液中,然后加入pyaop(2.5当量,22mg)并混合6小时。加入额外的pyaop(1.0当量,9mg)和diea(2.5当量,9μl),并混合12小时。加入额外的pyaop(1.0当量,9mg)和diea(2.5当量,9μl),并混合6小时。将该反应溶液在搅拌下缓慢加入冷水中。经过滤收集沉淀的产物,并随后用水洗涤3次(3x10ml)。真空干燥产物,得到白色固体seq id no:48(90mg,69.8%产率)。uplc:81.9面积%。
[0465]
实施例64:seq id no:48的总体脱保护以生成seq id no:6
[0466]
总体脱保护使用以下方法进行:1)将4ml切割试剂混合物tfa/h2o/tips/dtt(0.925/0.025/0.025/0.025)加入r1,然后加入seq id no:48(180mg)。2)在20-30℃搅拌3小时。3)将该溶液倒入冷的mtbe(30ml)中。搅拌该混悬液0.5小时。4)经过滤器过滤,然后用mtbe洗涤(30ml)两次。5)减压干燥该湿滤饼直至恒重。6)得到180mg干燥的粗产物,hplc纯度为66.3%。
[0467]
实施例65:天然化学连接
[0468]
合成树脂化合物53:
[0469][0470]
将fmoc-肼-2-氯三苯甲基树脂(30.06g,25.5mmol)在300ml dcm中溶胀15分钟。将其用2x400ml dmf溶胀每次15分钟。用3x400ml 20%哌啶/dmf进行fmoc脱保护每次30分钟。然后将该树脂用5x400ml dmf洗涤。将fmoc-l-lys(alloc)-oh(34.63g,76.5mmol,3.0当量)和hbtu(29.17g,76.9mmol,3.02当量)溶于400ml dmf中。将n,n-二异丙基乙胺(27ml,155mmol,6.08当量)加入该氨基酸溶液。然后将该溶液加入树脂制剂xx,将其搅拌6小时。将该树脂用5x400ml dmf、然后用5x300ml dcm洗涤,该树脂在35℃于真空烘箱中干燥约16小时。定量nmr测定该树脂负载量为0.52mmol/g。
[0471]
实施例66:合成肽酰肼seq id no:50(化合物54)
[0472]
将约12.19g树脂化合物53(5.4mmol,负载值:0.44mmol/g)用3x120ml dmf溶胀每次15分钟。使用标准spps如前文所述合成肽酰肼seq id no:50。
[0473]
脱保护:4x100ml 20%v/v哌啶在dmf中的溶液,每次20分钟。
[0474]
偶联:使用3当量氨基酸、3当量oxyma和3.3当量dic用于氨基酸偶联。
[0475]
spps过程中,每次偶联和最后一次fmoc脱保护后将该树脂用5x120ml dmf洗涤,同时用n2混合5min。
[0476]
在肽酰肼合成的最后,将树脂用dcm洗涤,同时用n2混合。将树脂在肽合成器上干燥。
[0477]
alloc脱保护和侧链偶联:
[0478]
将树脂用5x120ml dcm洗涤并搅拌5分钟。在75ml dcm中制备钯(iv)(500mg,
0.43mmol,0.1当量)和苯基硅烷(0.7ml,5.7mmol,1.02当量)溶液。将其加入该树脂中,并搅拌20分钟。将其用5x120ml dcm洗涤,并每次搅拌5分钟。用pd(pph3)4和phsih3进行alloc脱保护,重复两次。
[0479]
将该树脂用5x120ml dmf洗涤,并每次搅拌5分钟。将tntu(3.95g,10.82mmol,2当量)溶于(s)-13-(叔丁氧基羰基)-36,36-二甲基-10,15,34-三氧代-3,6,35-三氧杂-9,14-二氮杂三十七烷酸(化合物25,27ml,0.29g/ml,7.873g,10.8mmol,2当量)的dmf溶液中,并用dmf将其调至75ml。将3.8ml of n,n-二异丙基乙胺(3.8ml,21.82mmol,4.0mmol)加入化合物25的溶液中,并搅拌10分钟。然后将其加入树脂中,并搅拌14小时。然后将树脂用5x120ml dmf(5min搅拌)、5x120ml dcm(5min搅拌)洗涤。将树脂在35℃下在真空烘箱中干燥约16小时。
[0480]
总体脱保护和切割:
[0481]
将用2.5%w/v二硫苏糖醇(dtt)、2.5%v/v水、2.5%v/v三异丙基硅烷(tips)和92.5%三氟乙酸(tfa)制备的250ml切割试剂混合物加入在500ml三颈圆底烧瓶的干燥树脂(22.2g)中,并搅拌约2.5小时。将树脂过滤,并用2x7.5ml tfa洗涤。将滤液倒入1.40l冷却的mtbe中,肽立即沉淀出现。将过滤烧瓶用2x5ml tfa洗涤,并倒入冷却的mtbe中。将其冷却至
–
20℃持续半小时,然后离心。然后用300ml mtbe洗涤该肽沉淀物两次并离心。将该肽沉淀在27℃在真空烘箱中干燥约16小时。干燥后得到约9.9g粗的seq id no:50。
[0482]
实施例67:硫酯seq id no:51(化合物55)的合成:
[0483]
将粗的肽酰肼(seq id no:50,3.65g,1.41mmol)溶于250ml的连接缓冲液(6m盐酸胍和0.1m磷酸二氢钠溶液,ph约7.0)中。用5n hcl溶液将ph值调节至约3.3,并在丙酮-冰浴中将该溶液冷却至
–
15℃。将2.5ml 4.31m亚硝酸钠溶液(742.7mg,10.8mmol,7.6当量)加入该肽酰肼溶液中,并在
–
15℃下搅拌15分钟。同时,将4-巯基苯酚(1.052g,8.34mmol)悬浮于3ml连接缓冲液中,用5n naoh溶液将ph调至约7.0,并用连接缓冲液(6m盐酸胍和0.1m磷酸二氢钠缓冲液,ph约7.0)调至10ml。15分钟后,将7.5ml 4-巯基苯酚加入该肽酰肼溶液中,以引起从seq id no:50生成的肽基叠氮化物原位硫解。
[0484]
用5n氢氧化钠溶液将反应混合物的ph调至约6.5。将肽基叠氮化物的硫解进行15分钟,并将该粗的硫酯混合物经rp-hplc在phenomenex luna c18 10μm柱(30mm x 250mm)上在环境温度下纯化,整个纯化过程中用含有恒定5%乙酸铵的以下线性梯度洗涤:首先10%乙腈水溶液3分钟、然后10-30%乙腈水溶液3-5分钟、随后30-55%乙腈水溶液25分钟。得到约0.415g肽硫酯(seq id no:51)[预期值(质量 2h
)/2=1342.7052,实测值(质量 2h
)/2=1342.6958]。
[0485]
实施例68:天然化学连接以合成seq id no:53
[0486]
6m盐酸胍和0.1m磷酸二氢钠的水溶液(ph约7.0)是天然化学连接中使用的连接缓冲液。将该缓冲液用氮气脱气15分钟。将4-巯基苯酚(193mg,1.5mmol,10当量)、三(2-羧基乙基)膦(tcep,656.6mg,2.3mmol,15.3当量)和抗坏血酸(269mg,1.5mmol,10当量)放进一个三颈圆底烧瓶中。将烧瓶置于氮气下。加入41ml连接缓冲液以溶解在圆底烧瓶中的试剂。使用5n naoh溶液将溶液的ph值调至约7.0。将肽硫酯seq id no:51((406.8mg,0.15mmol)和n-末端半胱氨酸片段seq id no 52(化合物56)(326.5mg,0.15mmol,1当量)加到上述溶液中。用5n naoh溶液将ph调至约7.0。将反应混合物在氮气中搅拌约10小时。消耗了大部分
硫酯seq id no:51,因此将反应混合物在冰箱中于
–
20℃下储存约14小时。将seq id no:53经rp-hplc在phenomenex luna c18 10μm柱(30mm x 250mm)上在环境温度下纯化,整个纯化过程中用含有恒定5%乙酸铵的以下线性梯度洗涤:首先20%乙腈水溶液3分钟、然后20-30%乙腈水溶液3-5分钟、随后30-50%乙腈水溶液25分钟。得到约0.52g半胱氨酸类似物seq id no:53[预期值(质量 3h
)/3=1587.8219,实测值(质量 3h
)/3=1587.8198]。
[0487]
光脱硫:3m盐酸胍和0.1m磷酸二氢钠的水性缓冲液(ph约7.0)是新制备的。在该缓冲液中制备0.5ml 7.64mm三(2,2
′‑
联吡啶)二氯钌(ii)六水合物(2.86mg,0.004mmol)溶液。将三(2-羧基乙基)膦(tcep,64.3mg,0.22mmol)悬浮于该缓冲液中,并用5n naoh溶液将ph调至约7.0。将其用该缓冲液稀释至2ml。在7ml闪烁瓶中将seq id no:53(10mg,0.0021mmol)溶于4ml该缓冲液。将三苯基膦-3,3',3
”‑
三磺酸三钠盐(tppts,231.2mg,0.41mmol,194当量)和2-巯基乙磺酸钠盐(mesna,32.6mg,0.20mmol,95当量)加入seq id no:6的溶液中。将28μl三(2,2'-联吡啶)二氯钌(ii)六水合物(0.00021mmol,0.1当量)和20μl tcep溶液(0.0022mol,1.0当量)加入反应混合物中。将瓶子置于penn optical coating光反应器m1中,并以459rpm搅拌,led强度91%。风扇以3564rpm的转速运行,以防止反应混合物受热。约3.5小时后,进一步加入tcep(0.40mg,0.7当量),并在光反应器中搅拌反应混合物16小时。16小时后,反应完成,超过95%的seq id no:53转化为seq id no:6。
[0488]
无金属脱硫:6m盐酸胍和0.1m磷酸二氢钠(ph 7.0)的水溶液是用于该反应的缓冲液。用氮气彻底冲扫该缓冲液一小时以上。将seq id no:53(40.3mg,0.0085mmol)、2,2'-偶氮二[2-(2-咪唑啉-2-基)丙烷]二盐酸化物(27.7mg,0.0857mmol,10.1当量)、l-还原型谷胱甘肽(l-gsh,25.9mg,0.0843mmol,10当量)和三(2-羧基乙基)膦(tcep,36.5mg,0.1273mmol,15.0当量)溶于4ml缓冲液中。将反应混合物再次用氮气脱气约2分钟,并用5n naoh将ph调至约7.0。将该溶液在氮气中于45℃搅拌12小时,然后在室温下搅拌8小时。20小时后,大部分seq id no:53转化为seq id no:6。[预期值(质量 3h
)/3=1604.5153,实测值(质量 3h
)/3=1577.1581]。
[0489]
实施例69:使用seq id no 52和54用半胱氨酰丙酯(cpe)经天然化学连接(ncl)合成seq id no:53(化合物57)
[0490]
3m缓冲溶液:盐酸胍(2.86g,30.0mmol)、磷酸二氢钠(0.24g,2.0mmol)和盐酸三(2-羧基乙基)膦[tcep](0.0166g,0.0579mmol)称重加入15ml离心管内,溶于去离子水,并调至约9.5ml。按需加入5n naoh将缓冲溶液的ph调至~8.3。如果ph过头,则通过加入1n hcl再调至~8.3。
[0491]
连接的通用方法:
[0492]
将1ml缓冲溶液加入装有预先称重的seq id no:54[cpe-肽类似物](0.01g,0.003mmol,94.69质量%)和seq id no:52[cys-肽](0.0073g,0.0032mmol,96.9质量%)的5ml闪烁瓶中。经超声处理将肽片段完全溶于3m缓冲溶液中。记录该溶液的ph,并通过加入5n naoh调至ph~8.30。如果ph过头,通过加入1n hcl再调至~8.3。然后将溶液转移至hplc瓶中,并经eltivo在37℃(32℃内部温度)在不同时间点监测样品。通常在18小时后观察到完全转化为seq id no:53类似物(通过q-tof测定高达69%)。
[0493]
5m缓冲溶液:将盐酸胍(4.78g,50.0mmol)、磷酸二氢钠(0.24g,2.0mmol)和盐酸三(2-羧基乙基)膦[tcep](0.0166g,0.0579mmol)称重后加入15ml离心管中,溶于去离子水,
dmf洗涤1分钟,并用5x10ml二氯甲烷洗涤2分钟,然后干燥至恒重,以提供1.719g在树脂上的标题化合物。
[0504]
用由92.5%tfa、2.5%三异丙基硅烷、2.5%水和2.5%二硫苏糖醇(v/v/v/w)构成的2.0ml溶液从树脂中切割50mg肽样品。将该混合物在旋转混合器上搅拌1.5小时,用16ml 80:20dmso/乙腈(v/v)稀释,并经过滤以去除树脂。滤液经lc/ms分析,显示包含75.5%面积%的所需三肽和16.8%的含有多种乙醇酸添加物的产物。
[0505]
实施例71:fmoc-l-pro-乙醇酸-l-val-oh的替代合成
[0506]
步骤1(fmoc-l-val-oh偶联):
[0507]
在第一次偶联之前,将rink酰胺am树脂(0.74g/mmol,1.35g,1.00mmol)加入反应容器中。将该树脂用3x10ml dmf溶胀每次20分钟,然后用3x10ml 20%哌啶/dmf(v/v)脱保护每次30分钟,并用5x10ml dmf洗涤每次1分钟。制备(2s)-2-(9h-芴-9-基甲氧基羰基氨基)-3-甲基-丁酸(1.018g,3.00mmol)和1-羟基苯并三唑水合物(353mg,2.31mmol)在10ml dmf中的溶液。向该溶液中加入n,n'-二异丙基碳二亚胺(517μl,3.30mmol),并将对应的溶液加入装有溶胀树脂的反应容器中。将反应物在环境温度下混合4小时,然后将液体排空。将树脂用5x10ml dmf洗涤每次1分钟,然后进行下一步骤。
[0508]
步骤2(fmoc-乙醇酸偶联):
[0509]
通过用3x10ml 20%哌啶/dmf(v/v)处理来自步骤1的树脂每次30分钟并用5x10ml dmf洗涤每次1分钟来除去fmoc基团。制备2-(9h-芴-9-基甲氧基羰基氧基)乙酸(894.9mg,3.00mmol)和1-羟基苯并三唑水合物(353mg,2.31mmol)在10ml dmf中的溶液。向该溶液中加入n,n'-二异丙基碳二亚胺(517μl,3.30mmol),并将对应的溶液加入装有树脂的反应器中。将反应物在环境温度下混合16小时,然后将液体排空。将树脂用5x10ml dmf洗涤每次1分钟,然后进行下一步骤。
[0510]
步骤3(fmoc-l-pro-oh偶联):
[0511]
通过用3x10ml 20%哌啶/dmf(v/v)各处理30分钟并用5x10ml dmf洗涤每次1分钟来除去fmoc基团。制备(2s)-1-(9h-芴-9-基甲氧基羰基)吡咯烷-2-甲酸(1.012g,3.00mmol)、(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐(hbtu)(1.25g,3.30mmol)和n,n-二异丙基乙胺(870μl,5.00mmol)在10ml dmf中的溶液。将对应的溶液加入装有树脂的反应器中。反应物在环境温度下混合8小时,然后排空。将树脂用5x10ml dmf洗涤每次1分钟,用5x10ml二氯甲烷洗涤1分钟,然后干燥至恒重,以提供1.622g在树脂上的标题化合物。
[0512]
用由92.5%tfa、2.5%三异丙基硅烷、2.5%水和2.5%二硫苏糖醇(v/v/v/w)构成的2.0ml溶液从树脂中切割50mg肽样品。将该混合物在旋转混合器上搅拌1.5小时,用16ml 80:20dmso/乙腈(v/v)稀释,并经过滤以去除树脂。滤液经lc/ms分析,显示包含84.93面积%的所需三肽,没有可检测到的多种乙醇酸添加物。
[0513]
实施例72:2-(9h-芴-9-基甲氧基羰基氧基)乙酸(fmoc-乙醇酸)(化合物59)合成
[0514][0515]
步骤1(2-(9h-芴-9-基甲氧基羰基氧基)乙酸叔丁酯):
[0516]
向500ml圆底烧瓶中的2-羟基乙酸叔丁酯(10.00g,71.90mmol,95质量%)在120ml二氯甲烷中的磁力搅拌的溶液中一次性加入吡啶(60ml,742.0mmol)。将所得溶液在冰浴中冷却至0-5℃。经滴液漏斗历经30分钟向该溶液中逐滴加入9-芴甲基氯甲酸酯(20.00g,77.30mmol)在60ml二氯甲烷中的溶液。当加完时,反应物中已形成沉淀。移除冰浴,并将反应混合物在环境温度下搅拌18小时。在额外的搅拌时间内形成更多的沉淀物。将反应混合物在减压下浓缩成固体油状残余物,以去除大部分吡啶和二氯甲烷,然后重新溶解在200ml二氯甲烷中。用2x100ml 1m硫酸氢钠水溶液和2x100ml饱和盐水溶液先后洗涤该溶液。将有机层在硫酸镁上干燥,浓缩成27.13克的黄色油状物,其逐渐凝固。粗产物不经纯化直接在下一步骤中进行处理。
[0517]
步骤2(2-(9h-芴-9-基甲氧基羰基氧基)乙酸):
[0518]
将来自步骤1的2-(9h-芴-9-基甲氧基羰基氧基)乙酸叔丁酯(26.0g,73.40mmol)溶于二氯甲烷(200ml)。向该磁力搅拌的溶液中加入三氟乙酸(52ml),然后加入三异丙基硅烷(13ml)。将所得溶液在环境温度下搅拌3小时。减压浓缩该溶液以除去二氯甲烷和几乎所有的三氟乙酸。用1000ml 5%碳酸氢钠水溶液逐渐处理所得粘性残余物,以防止起泡,并用3x500ml甲基叔丁基醚洗涤该水溶液,以去除残留的三异丙基硅烷。将水溶液冷却至0-5℃,并加入300ml乙酸乙酯。将该两相混合物用40%磷酸水溶液酸化至~ph 2,需要约75ml酸。分离各层后,将有机层在硫酸镁上干燥,并减压浓缩成粘稠的淡黄色油状物。将该油状物在冰箱中冷却至-20℃,使该物质完全固化为白色固体。将该固体用75ml冷却的庚烷研磨,并超声处理后,形成均匀的白色混悬液。过滤固体,用庚烷洗涤,并在真空烘箱中于33℃下干燥过夜,得到15.21g(历经两个步骤的产率为69.5%)的白色固体。
[0519]
nmr(cdcl3)确认所需产物已被分离。
[0520]
实施例73:fmoc-lys(mtt)-cys(trt)-oh(化合物60)合成
[0521][0522]
步骤1
[0523]
将(2s)-6-[[二苯基(对甲苯基)甲基]氨基]-2-(9h-芴-9-基甲氧基羰基氨基)己酸(a,15g,24.01mmol)、n,n'-二琥珀酰亚胺基碳酸酯(7.45g,29.0mmol,99.6质量%)、4-二甲基氨基吡啶[dmap](0.3g,2mmol,99质量%)称重装入配有搅拌棒的250ml烧瓶中。然后加入乙酸乙酯(225ml,2000mmol,100质量%),并将该溶液在室温(21-24℃)下混合直至获得溶液。将反应物搅拌18小时或直至经lcms/nmr确认反应完成。将反应混合物转移至分液漏斗,用去离子水(60ml x3)洗涤,并将有机层在旋转蒸发仪上浓缩至干,得到粗的步骤1的化合物。
[0524]
步骤2
[0525]
向步骤1的粗的化合物(17.33g,24.01mmol)在n,n-二甲基甲酰胺(208ml,2690mmol)的溶液中加入n,n-二异丙基乙胺(5.03ml,28.8mmol)和(2r)-2-氨基-3-三苯甲基硫基-丙酸(9.6g,26mmol)。在室温(21-24℃)下使用磁力搅拌将反应物搅拌18小时或直至经lcms/nmr确认反应完成。将反应混合物转移到分液漏斗中,用10%柠檬酸(120ml x 2)洗涤,并用二氯甲烷(100ml x 5)萃取。将有机层用去离子水(100ml x 2)洗涤,并在48
–
50℃的旋转蒸发仪上将合并的有机层浓缩至干,以去除多余的溶剂。将粗的化合物60在超声
处理下溶于乙腈(30ml)。将粗的化合物60溶液在搅拌中逐滴加入冷却的乙腈:去离子水(3:2,700ml)中。将该浆体在0℃搅拌过夜。过滤固体,用己烷(70ml)洗涤,并在真空烘箱中以40℃干燥,得到产物fmoc-lys(mtt)-cys(trt)-oh(化合物60)(21.0g,76.8%产率,通过q-nmr校正效能)。
[0526]
实施例74:fmoc-l-lys(mtt)-l-cys(trt)-l-pro-乙醇酸-l-val-oh(化合物61)合成
[0527][0528]
在偶联反应之前,将来自上述实施例73的树脂上的fmoc-l-pro-乙醇酸-l-val-oh(1.719g,1.00mmol)用3x15ml dmf溶胀每次20分钟,然后用4x15ml 20%哌啶/dmf(v/v)脱保护每次30分钟,并用5x15ml dmf洗涤每次1分钟。制备(2r)-2-[[(2s)-6-[[二苯基(对甲苯基)甲基]氨基]-2-(9h-芴-9-基甲氧基羰基氨基)己酰基]氨基]-3-三苯甲基硫基-丙酸(2.28g,2.00mmol,85.24质量%)、羟基-3,4-二氢-4-氧代-1,2,3-苯并三嗪(hoobt)(0.375g,2.30mmol,95质量%)和n,n'-二异丙基碳二亚胺(0.41ml,2.60mmol)在10ml dmf中的溶液。将相应的溶液加至装有树脂的反应器中。将反应物在环境温度下混合12小时,并将液体排空。将树脂用5x15ml dmf洗涤每次1分钟,并用5x15ml二氯甲烷洗涤每次1分钟,然后干燥至恒重,以提供1.973g在树脂上的标题化合物。
[0529]
用由92.5%tfa、2.5%三异丙基硅烷,2.5%水和2.5%二硫苏糖醇(v/v/v/w)构成的2.0ml溶液从树脂中切割50mg肽样品。将该混合物在旋转混合器上搅拌1.5小时,用16ml 80:20dmso/乙腈(v/v)稀释,并经过滤以去除树脂。滤液经lc/ms分析,显示包含81.93%面积%所需的五肽以及2.91面积%的去甲基脯氨酸和3.83%的去甲基缬氨酸。
[0530]
实施例75:
[0531]
fmoc-gly-pro-ser(tbu)-ser(tbu)-gly-ala-pro-pro-pro-ser(tbu)-oh(seq id no:55)合成
[0532]
标题化合物如下文所述使用标准固相合成条件(fmoc-保护的氨基酸/乙基氰基乙醛酸-2-肟(oxyma)/n,n
’‑
二异丙基碳二亚胺(dic)制备。
[0533]
溶剂和试剂制备:
[0534]
将20l dmf加入溶剂储器中。将5l 20%哌啶/dmf(v/v)溶液加入该脱保护的储器中。使用n,n'-二异丙基碳二亚胺(49.98g,396.0mmol)和dmf制备600ml 0.660m dic溶液,并加入dic/溶剂储器中。使用乙基氰基乙醛酸-2-肟(53.29g,371.2mmol)和dmf制备500ml 0.750m oxyma溶液,并加入oxyma/溶剂储器中。将sieber树脂(0.71mmol/g,14.09g,10.00mmol)加入反应器中。如下所示的合成步骤开始前,将树脂用3x180ml dmf溶胀每次20分钟,并用3x180ml 20%哌啶/dmf(v/v)去除fmoc基团每次30分钟。
[0535]
氨基酸溶液制备:
[0536]
从(2s)-2-(9h-芴-9-基甲氧基羰基氨基)丙酸(11.68g,37.52mmol)和dmf制备100ml 0.375m fmocnh-l-ala-oh溶液,并加入合适的氨基酸瓶中。从2-(9h-芴-9-基甲氧基羰基氨基)乙酸(22.30g,75.01mmol)和dmf制备200ml0.375m fmocnh-gly-oh溶液,并加入合适的氨基酸瓶中。从(2s)-1-(9h-芴-9-基甲氧基羰基)吡咯烷-2-甲酸(45.54g,135.0mmol)和dmf制备360ml 0.375m fmocnh-l-pro-oh溶液,并加入合适的氨基酸瓶中。从(2s)-3-叔丁氧基-2-(9h-芴-9-基甲氧基羰基氨基)丙酸(40.25g,105.0mmol)和dmf制备280ml 0.375m fmocnh-l-ser(tbu)-oh溶液,并加入合适的氨基酸瓶中。
[0537]
偶联条件:
[0538]
pro:0.18m,3.0当量氨基酸,3.0当量oxyma/3.3当量dic,活化的酯溶液预活化30分钟,在环境温度下偶联时间6小时,用20%哌啶/dmf(v/v)4x30分钟脱保护,脱保护后及偶联后进行5x2分钟dmf洗涤。
[0539]
ala(pro后)、gly(pro后):0.18m,3.0当量氨基酸,3.0当量oxyma/3.3当量dic,活化的酯溶液预活化30分钟,在环境温度下偶联时间4小时,用20%哌啶/dmf(v/v)4x30分钟脱保护,脱保护后及偶联后进行5x2分钟dmf洗涤。
[0540]
全部其它偶联:0.18m,3.0当量氨基酸,3.0当量oxyma/3.3当量dic,活化的酯溶液预活化30分钟,在环境温度下偶联时间4小时,用20%哌啶/dmf(v/v)3x30分钟脱保护,脱保护后及偶联后进行5x2分钟dmf洗涤。
[0541]
在合成最后,将树脂用5x180ml dmf各洗涤2分钟,然后用5x180ml mtbe各洗涤2分钟。将树脂从反应器中移出,转移至配衡的结晶盘,并在40℃真空干燥至恒重,以提供在树脂上的25.15g标题化合物。基于树脂起始材料的质量,肽的产率为11.07g(89%)。
[0542]
通过用3x6ml dmf溶胀树脂10分钟,用3x6ml 20%哌啶/dmf(v/v)各处理30分钟,用5x6ml dmf洗涤每次1分钟,用5x6ml二氯甲烷洗涤每次1分钟并干燥至恒重,从而从251mg树脂上的肽样品中去除fmoc基团。
[0543]
通过用5ml tfa/tis/h2o/dtt([0.925v:0.025v:0.025v]:0.025w)溶液在20ml闪烁瓶中在旋转混合器上混合2小时,从树脂上切割该脱保护的产物样品。滤出树脂,并将树脂的湿滤饼用2ml纯tfa洗涤。
[0544]
将所得粗的肽用35ml冷却的mtbe沉淀,离心,用2x35ml mtbe洗涤,并在33℃真空干燥过夜,得到105.1mg(94.9%)全部脱保护的肽。uplc分析显示98.62面积%纯度,没有相关物质超过0.30面积%。肽在树脂上的负载量检测为0.37mmol/g,而理论负载量为
0.37mmol/g。
[0545]
实施例75:
[0546]
h-cys-gln-aib-phe-ile-glu-tyr-leu-leu-glu-gly-gly-pro-ser-ser-gly-ala-pro-pro-pro-ser-nh2seq id no:52(化合物62)合成
[0547]
标题化合物如下文所述使用标准固相合成条件(fmoc-保护的氨基酸/乙基氰基乙醛酸-2-肟(oxyma)/n,n
’‑
二异丙基碳二亚胺(dic)制备。
[0548]
溶剂和试剂制备:
[0549]
将40l dmf加入溶剂储器中。将4l 20%哌啶/dmf(v/v)溶液加入该脱保护的储器中。使用n,n'-二异丙基碳二亚胺(49.98g,396.0mmol)和dmf制备600ml 0.660m dic溶液,并加入dic/溶剂储器中。使用乙基氰基乙醛酸-2-肟(53.29g,371.2mmol)和dmf制备500ml 0.750m oxyma溶液,并加入oxyma/溶剂储器中。将sieber树脂(0.41mmol/g,1.22g,0.500mmol)上的9h-芴-9-基甲基n-[2-[(2s)-2-[[(1s)-2-[[(1s)-2-[[2-[[(1s)-2-[(2s)-2-[(2s)-2-[(2s)-2-[[(1s)-2-氨基-1-(叔丁氧基甲基)-2-氧代-乙基]氨甲酰基]吡咯烷-1-羰基]吡咯烷-1-羰基]吡咯烷-1-基]-1-甲基-2-氧代-乙基]氨基]-2-氧代-乙基]氨基]-1-(叔丁氧基甲基)-2-氧代-乙基]氨基]-1-(叔丁氧基甲基)-2-氧代-乙基]氨甲酰基]吡咯烷-1-基]-2-氧代-乙基]氨基甲酸酯加入11个反应器各自当中(共5.5mmol树脂上的肽)。如下所示的合成步骤开始前,将各反应器中的树脂用3x10ml dmf溶胀每次20分钟,然后用3x10ml 20%哌啶/dmf(v/v)去除fmoc基团各30分钟,并将树脂用5x10ml dmf洗涤每次1分钟。
[0550]
氨基酸溶液制备:
[0551]
1. 57ml 0.375m fmocnh-l-ala-oh溶液是从(2s)-2-(9h-芴-9-基甲氧基羰基氨基)丙酸(6.66g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0552]
2. 103ml 0.375m fmocnh-l-glu(tbu)-oh溶液是从(2s)-5-叔丁氧基-2-(9h-芴-9-基甲氧基羰基氨基)-5-氧代-戊酸(16.44g,38.63mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0553]
3. 61ml 0.375m fmocnh-l-phe-oh溶液是从(2s)-2-(9h-芴-9-基甲氧基羰基氨基)-3-苯基-丙酸(8.83g,22.79mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0554]
4. 57ml 0.375m fmocnh-gly-oh溶液是从2-(9h-芴-9-基甲氧基羰基氨基)乙酸(6.36g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0555]
5. 57ml 0.375m fmocnh-l-ile-oh溶液是从(2s,3s)-2-(9h-芴-9-基甲氧基羰基氨基)-3-甲基-戊酸(7.55g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0556]
6. 103ml 0.375m fmocnh-l-leu-oh溶液是从(2s)-2-(9h-芴-9-基甲氧基羰基氨基)-4-甲基-戊酸(13.65g,38.62mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0557]
7. 82ml 0.375m fmocnh-l-gln(trt)-oh溶液是从(2s)-5-(叔丁基氨基)-2-(9h-芴-9-基甲氧基羰基氨基)-5-氧代-戊酸(13.05g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0558]
8. 57ml 0.375m fmocnh-l-tyr(tbu)-oh溶液是从(2s)-3-(4-叔丁氧基苯基)-2-(9h-芴-9-基甲氧基羰基氨基)丙酸(9.82g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
val-nh2(seq id no:56;化合物63)合成
[0570]
标题化合物使用标准固相合成条件(fmoc-保护的氨基酸/乙基氰基乙醛酸-2-肟(oxyma)/n,n
’‑
二异丙基碳二亚胺(dic)制备。
[0571]
溶剂和试剂制备:
[0572]
将40l dmf加入溶剂储器中。将4l 20%哌啶/dmf(v/v)溶液加入该脱保护的储器中。使用n,n'-二异丙基碳二亚胺(49.98g,396.0mmol)和dmf制备600ml 0.660m dic溶液,并加入dic/溶剂储器中。使用乙基氰基乙醛酸-2-肟(53.29g,371.2mmol)和dmf制备500ml 0.750m oxyma溶液,并加入oxyma/溶剂储器中。将rink amide am、rink amide mbha或sieber树脂(0.500mmol)上的[2-[[(1s)-1-氨甲酰基-2-甲基-丙基]氨基]-2-氧代-乙基](2s)-1-[(2r)-2-[[(2s)-6-[[二苯基(对甲苯基)甲基]氨基]-2-(9h-芴-9-基甲氧基羰基氨基)己酰基]氨基]-3-三苯甲基硫基-丙酰基]吡咯烷-2-甲酸酯加入8个反应器各自当中(共4.0mmol树脂上的肽)。
[0573]
如下所示的合成步骤开始前,将各反应器中的树脂用3x10ml dmf溶胀每次20分钟,然后用4x10ml 20%哌啶/dmf(v/v)去除fmoc基团各30分钟,并将树脂用5x10ml dmf洗涤每次1分钟。
[0574]
氨基酸溶液制备:
[0575]
1. 57ml 0.375m fmocnh-aib-oh溶液是从2-(9h-芴-9-基甲氧基羰基氨基)-2-甲基-丙酸(6.96g,21.37mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0576]
2. 103ml 0.375m fmocnh-l-asp(tbu)-oh溶液是从(2s)-4-叔丁氧基-2-(9h-芴-9-基甲氧基羰基氨基)-4-氧代-丁酸(15.87g,38.63mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0577]
3. 57ml 0.375m fmocnh-l-phe-oh溶液是从(2s)-2-(9h-芴-9-基甲氧基羰基氨基)-3-苯基-丙酸(8.28g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0578]
4. 57ml 0.375m fmocnh-gly-oh溶液是从2-(9h-芴-9-基甲氧基羰基氨基)乙酸(6.36g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0579]
5. 57ml 0.375m fmocnh-l-ile-oh溶液是从(2s,3s)-2-(9h-芴-9-基甲氧基羰基氨基)-3-甲基-戊酸(7.55g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0580]
6. 57ml 0.375m fmocnh-l-lys(boc)-oh溶液是从(2s)-6-(叔丁氧基羰基氨基)-2-(9h-芴-9-基甲氧基羰基氨基)己酸(10.02g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0581]
7. 57ml 0.375m fmocnh-l-leu-oh溶液是从(2s)-2-(9h-芴-9-基甲氧基羰基氨基)-4-甲基-戊酸(7.55g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0582]
8. 57ml 0.375m fmocnh-l-gln(trt)-oh溶液是从(2s)-5-(叔丁基氨基)-2-(9h-芴-9-基甲氧基羰基氨基)-5-氧代-戊酸(13.05g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0583]
9. 103ml 0.375m fmocnh-l-ser(tbu)-oh溶液是从(2s)-3-叔丁氧基-2-(9h-芴-9-基甲氧基羰基氨基)丙酸(14.81g,38.63mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0584]
10. 103ml 0.375m fmocnh-l-thr(tbu)-oh溶液是从(2s,3r)-3-叔丁氧基-2-(9h-芴-9-基甲氧基羰基氨基)丁酸(15.35g,38.63mmol)和dmf制备,并加入合适的氨基酸
瓶中。
[0585]
11. 57ml 0.375m fmocnh-l-tyr(tbu)-oh溶液是从(2s)-3-(4-叔丁氧基苯基)-2-(9h-芴-9-基甲氧基羰基氨基)丙酸(9.82g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0586]
12. 57ml 0.375m bocnh-l-tyr(tbu)-oh溶液是从(2s)-2-(叔丁氧基羰基氨基)-3-(4-叔丁氧基苯基)丙酸(7.21g,21.38mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0587]
13. 57ml 0.375m fmocnh-l-αmeleu-oh溶液是从(2r)-2-(9h-芴-9-基甲氧基羰基氨基)-2,4-二甲基-戊酸(7.85g,21.37mmol)和dmf制备,并加入合适的氨基酸瓶中。
[0588]
偶联条件:
[0589]
boc-tyr、ile:0.18m,3.0当量氨基酸,3.0当量oxyma/3.3当量dic,活化的酯溶液预活化30分钟,在环境温度下18小时偶联时间,用20%哌啶/dmf(v/v)4x30分钟脱保护,脱保护后及偶联后进行5x1分钟dmf洗涤。
[0590]
aib、gln、αmeleu:0.18m,3.0当量氨基酸,3.0当量oxyma/3.3当量dic,活化的酯溶液预活化30分钟,在环境温度下12小时偶联时间,用20%哌啶/dmf(v/v)4x30分钟脱保护,脱保护后及偶联后进行5x1分钟dmf洗涤。
[0591]
全部其它偶联:0.18m,3.0当量氨基酸,3.0当量oxyma/3.3当量dic,活化的酯溶液预活化30分钟,在环境温度下4小时偶联时间,用20%哌啶/dmf(v/v)3x30分钟脱保护,脱保护后及偶联后进行5x1分钟dmf洗涤。在合成最后,将树脂用5x10ml dmf洗涤每次1分钟,然后用5x1ml二氯甲烷洗涤每次1分钟。将树脂干燥至恒重,并进行下一步骤。用5ml tfa/tis/h2o/dtt([0.925v:0.025v:0.025v]:0.025w)溶液在20ml闪烁瓶中在旋转混合器上混合2小时,以从树脂上切割来自反应器之一中的样品(~80mg)。滤出树脂,并将树脂的湿滤饼用2ml纯tfa洗涤。将所得粗的肽用35ml冷却的mtbe沉淀,离心,用2x35ml mtbe洗涤,并在33℃真空干燥过夜,得到全部脱保护的肽的样品。经uplc分析显示59.8面积%纯度。
[0592]
实施例77:
[0593]
tyr-aib-gln-glu-thr-phe-thr-ser-asp-tyr-ser-ile-αmeleu-leu-asp-lys(aeea-aeea-γglu-c
20-oh)-cys-pro-乙醇酸-val-nh2(seq id no:57;化合物64)合成
[0594]
步骤1(mtt保护基团的脱保护):
[0595]
将在rink amide am、rink amide mbha或sieber树脂(0.500mmol)上的[2-[[(1s)-1-氨甲酰基-2-甲基-丙基]氨基]-2-氧代-乙基](2s)-1-[(2r)-2-[[(2s)-2-[[(2s)-2-[[(2s)-4-叔丁氧基-2-[[(2s)-2-[[(2s)-2-[[(2s,3s)-2-[[(2s)-3-叔丁氧基-2-[[(2s)-2-[[(2s)-4-叔丁氧基-2-[[(2s)-3-叔丁氧基-2-[[(2s,3r)-3-叔丁氧基-2-[[(2s)-2-[[(2s,3r)-3-叔丁氧基-2-[[2-[[(2s)-2-[[2-[[(2s)-2-(叔丁氧基羰基氨基)-3-(4-叔丁氧基苯基)丙酰基]氨基]-2-甲基-丙酰基]氨基]-5-氧代-5-(三苯甲基氨基)戊酰基]氨基]乙酰基]氨基]丁酰基]氨基]-3-苯基-丙酰基]氨基]丁酰基]氨基]丙酰基]氨基]-4-氧代-丁酰基]氨基]-3-(4-叔丁氧基苯基)丙酰基]氨基]丙酰基]氨基]-3-甲基-戊酰基]氨基]-2,4-二甲基-戊酰基]氨基]-4-甲基-戊酰基]氨基]-4-氧代-丁酰基]氨基]-6-(叔丁氧基羰基氨基)己酰基]氨基]-6-[[二苯基(对甲苯基)甲基]氨基]己酰基]氨基]-3-三苯甲基硫基-丙酰基]吡咯烷-2-甲酸酯分别加入8个不同反应器中。将各树脂用3x10ml dcm各溶胀15分钟,然后用1,1,1,3,3,3-六氟-2-丙醇的30%在二氯甲烷中的溶液(v/v)
(10ml,94.98mmol)处理并混合1小时。将液体排空,并将树脂再次用1,1,1,3,3,3-六氟-2-丙醇的30%在二氯甲烷中的溶液(v/v)(10ml,94.98mmol)处理并混合1小时。将液体再次排空,并将树脂用5x10ml二氯甲烷洗涤每次1分钟,然后用5x10ml dmf洗涤每次1分钟,并直接进行偶联反应。
[0596]
步骤2(侧链的偶联):
[0597]
将2-[2-[2-[[(4s)-5-叔丁氧基-4-[(20-叔丁氧基-20-氧代-花生酰基)氨基]-5-氧代-戊酰基]氨基]乙氧基]乙氧基]乙酸(6.41g,8.00mmol,91质量%)和苯并三唑-1-基氧基三吡咯烷子基鏻六氟磷酸盐(pybop)(4.16g,8.00mmol)溶于72ml dmf中。加入n,n-二异丙基乙胺(1.40ml,8.00mmol),并将所得溶液振摇1分钟,然后将该溶液的八分之一加入各反应容器的树脂中,并混合16小时。将液体排空,并将树脂用5x15ml dmf洗涤每次1分钟,5x15ml二氯甲烷洗涤每次1分钟,然后干燥至恒重,得到15.66g树脂上的肽。
[0598]
步骤3(从树脂上切割肽与总体脱保护):
[0599]
通过机械搅拌用由148ml三氟乙酸、4.0ml三异丙基硅烷、4.0ml水和4.0g二硫苏糖醇构成的160ml溶液在环境温度下在三颈圆底烧瓶中从树脂上切割肽持续2小时。通过在釉料漏斗上过滤移出树脂,并用64ml tfa洗涤,得到总溶液体积约224ml。通过加入到1120ml冷却的mtbe中沉淀肽。在-20℃下老化1小时后,将该浆体分成四个瓶子并离心。将离心后所得固体合并到两个瓶子中,并将每份固体用250ml室温的mtbe洗涤两次。将所得固体在真空烘箱中于33℃干燥过夜,得到7.817g粗的标题化合物。
[0600]
实施例78:seq id no:6的纯化
[0601]
在5l反应器中将粗产物(76.23g)溶于3.05l 25%acn/水混合物(25g/l粗物质浓度)并搅拌30分钟。通过使用28%氢氧化铵调整ph=9.0转化并搅拌60分钟,然后再调整回酸性侧(使用tfa调至ph=2),以矫正缩酚酸肽异构体。最终acn含量需要为30%,以确保ph值调整后的溶解性。在第一个色谱步骤前过滤该粗物质。
[0602]
第一个色谱步骤:柱:dac200,200mm x 250mm,固定相(ymc triart c18,10um,12nm);流动相a:0.1%tfa在h2o中;流动相b:100%acn;在230nm检测;注射体积:3.5l(通过具有300ml/min流速的注射泵进行)。
[0603]
梯度:
[0604]
时间/min流速/(ml/min)%a%b01000703011000703052.510004555
[0605]
收集含有所需产物的流份。
[0606]
第二个色谱步骤:以等体积的h2o稀释,并用稀释的氨将ph调至6.5。柱:dac200,200mm x 250mm,固定相(ymc triart c18,10um,12nm)。流动相a:10mm nh4hco3在h2o中;流动相b:100%acn;在230nm检测;注射体积:7.0l(通过具有300ml/min流速的注射泵进行)。
[0607]
梯度:
[0608]
时间/min流速/(ml/min)%a%b010007030110007030
52.510004555
[0609]
收集含有所需产物的流份。
[0610]
钠盐转化步骤:将溶于200ml h2o中的1.76g(44.0mmol)naoh逐滴加入7.2l分离的流份中并冷冻干燥。得到38.02g纯化产物(纯度:98.0%)。
[0611]
实施例79:seq id no:29纯化
[0612]
粗产物用下述条件纯化:20cm柱(4.8kg daiso c18-ods-rps,10μ,)和流动相a:0.1%tfa在h2o中;流动相b:100%acn;在230nm检测。第一个纯化步骤:
[0613]
梯度:
[0614]
时间/min流速/(ml/min)%a%b0600802036007030756004555
[0615]
收集具有约88%或更高纯度的产物流份,用h2o 1:1稀释,用50%nh4oh将ph调至6.5。
[0616]
第二个色谱步骤:使用第一个步骤中的柱子。流动相a:10mm nh4hco3在h2o中;流动相b:100%acn;在230nm检测。
[0617]
梯度:
[0618]
时间/min流速/(ml/min)%a%b06008020456005050
[0619]
收集具有约97%纯度或更高纯度的流份。
[0620]
通过加入3当量naoh转化为钠盐,然后冷冻干燥。
[0621]
序列
[0622]
在本文中引用以下氨基酸序列,并在下面提供它们作为参考。
[0623]
seq id no:1
–
人gip
[0624]
yaegtfisdysiamdkihqqdfvnwllaqkgkkndwkhnitq
[0625]
seq id no:2
–
人glp-1(7-36)酰胺
[0626]
haegtftsdvssylegqaakefiawlvkgr-nh2[0627]
seq id no:3
–
人gcg
[0628]
hsqgtftsdyskyldsrraqdfvqwlmnt
[0629]
seq id no:4
–
肠促胰岛素类似物
[0630]
yx2qgtftsdysix
13
ldkx
17
ax
19
x
20
afieyllx
28
x
29
gpssx
34
appps,其中x2是aib,x
13
是l或αmel,x
17
是任何具有可用于偶联的官能团的氨基酸,并且所述官能团与c
16-c
22
脂肪酸偶联,x
19
是q或a,x
20
是aib、αmek、q或h,x
28
是e或a,x
29
是g或aib,x
34
是g或aib,且c-末端氨基酸任选地被酰胺化。
[0631]
seq id no:5
–
肠促胰岛素类似物
[0632]
y(aib)qgtftsdysi(αmel)ldkkaq(aib)afieylleggpssgappps
[0633]
seq id no:6
–
肠促胰岛素类似物
[0634]
y(aib)qgtftsdysi(αmel)ldkk((2-[2-(2-氨基-乙氧基)-乙氧基]-乙酰基)-(γglu)-co-(ch2)
18-co2h)aq(aib)afieylleggpssgappps-nh2[0635][0636]
seq id no:7
–
中间体化合物1
[0637][0638]
seq id no:8
–
中间体化合物2
[0639][0640]
seq id no:9
–
中间体化合物3
[0641][0642]
seq id no:10
–
中间体化合物4
[0643][0644]
seq id no:11
–
中间体化合物5
[0645][0646]
seq id no:12
–
中间体化合物6
[0647][0648]
seq id no:13
–
中间体化合物7
[0649][0650]
seq id no:14
–
中间体化合物8
[0651][0652]
seq id no:15
–
中间体化合物9
[0653][0654]
seq id no:16
–
中间体化合物10
[0655][0656]
seq id no:17
–
中间体化合物11
[0657][0658]
seq id no:18
–
中间体化合物12
[0659][0660]
seq id no:19
–
中间体化合物13
[0661][0662]
seq id no:20
–
中间体化合物14
[0663][0664]
seq id no:21
–
中间体化合物15
[0665][0666]
seq id no:22
–
中间体化合物16
[0667][0668]
seq id no:23
–
中间体化合物17
[0669][0670]
其中r可以是2,2,2-三氟乙基。
[0671]
seq id no:24
–
中间体化合物18
[0672][0673]
seq id no:25
–
中间体化合物19
[0674][0675]
其中r可以是2,2,2-三氟乙基。
[0676]
seq id no:26
–
中间体化合物20
[0677][0678]
seq id no:27
–
中间体化合物21
[0685][0686]
seq id no:30
–
中间体化合物27
[0687]
boc-y-aib-egt-αmef(2f)-tsd-4pal-si-αmel-l
[0688]
[0707][0708]
seq id no:38
–
中间体化合物23
[0709][0710]
seq id no:39
–
中间体化合物24
[0711][0712]
r是-ch
2-c(o)-val-nh2。
[0713]
seq id no:40
–
中间体化合物25
[0714][0715]
r是-ch
2-c(o)-val-nh2。
[0716]
seq id no:41
–
中间体化合物26
[0717][0718]
r是-ch
2-c(o)-val-nh2。
[0719]
seq id no:42
[0720][0721]
seq id no:43
[0722][0723]
seq id no:44
[0724][0725]
seq id no:45
[0726][0727]
seq id no:46
[0728][0729]
seq id no:47
[0730][0731]
seq id no:48
[0732][0733]
seq id no:49
[0734][0735]
seq id no:50
[0736][0737]
seq id no:51
[0738][0739]
seq id no:52
[0740]
cq-(aib)-afieylleggpssgappps-nh2[0741]
seq id no:53
[0742][0743]
seq id no:54
[0744]
[0745]
其中r是pro-乙醇酸-val或pro-乙醇酸。
[0746]
seq id no:55
[0747][0748]
seq id no:56
[0749][0750]
seq id no:57
[0751][0752]
seq id no:58
[0753][0754]
seq id no:59
[0755]
cq-(aib)-efi-(d-glu)-(α-甲基-tyr)-liegggpssgappps-nh2[0756]
seq id no:60
[0757][0758]
seq id no:61
[0759]
[0760]
seq id no:62
[0761]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。