1.本发明涉及药物化学合成技术领域,具体涉及一种硝基咪唑吡喃类药物的新合成工艺方法。
背景技术:
2.结核病是一种古老的传染病,是全球第九大致死疾病,耐药结核(dr-tb)的上升更加剧了结核病的蔓延。2016年,全球范围新增49万耐多药结核新增患者,包括一部分泛耐药结核患者。2017年,耐药结核患者估计超过55万,高度耐药患者尤其难以治愈,2015 年只有34%的耐多药结核患者成功获得治疗。
3.2016年,原sfda有条件批准将贝达喹啉作为联合治疗的一部分,用于成人耐多药结核的治疗。通过新药引入和保护机制,全国定点医疗机构使用贝达喹啉联合其他抗结核药物对耐多药患者进行规范的治疗管理。
4.普托马尼(pretomanid)是一种新型化合物,属于硝基咪唑类药物,与另一款抗结核新药德拉马尼(delamanid)一样均由日本大冢制药开发,非营利组织全球结核病药物研发联盟(tb alliance,简称结核联盟)2002年获得pretomanid的授权开始临床前开发。获得澳大利亚、德国、英国和美国等多国政府和慈善机构支持(参见:chen g,zhu m,cheny,et al.an efficient and practical protocol for the production of pretomanid(pa-824)via anovel synthetic strategy[j].chemical papers,2020,74:3937-3945.)。
[0005]
普托马尼是近半个世纪以来批准上市的第3个抗结核病新药,这是第一个由非营利组织研发和注册的抗结核新药。获得了fda的一系列优惠,包括优先审评资格、合格治疗传染病产品资格(qualified infectious disease product,qidp)及孤儿药资格。
[0006]
2019年8月14日,美国fda批准了抗结核新药普托马尼,与贝达喹啉和利奈唑胺组成bpal方案针对有限特定患者人群,即广泛耐药结核(xdr-tb)或无法耐受治疗/治疗欠佳的耐多药结核(mdr-tb)成人患者(参见:u.s.food and drug administration.fdaapproves new drug for treatment resistant forms of tuberculosis that affects the lungs[eb/ol]. [2019-08-14].https://www.fda.gov/news events/press announcements/fda approves new drugtreatment resistant forms tuberculosis affects lungs.)。
[0007]
在此之前,强生的耐多药结核药贝达喹啉于2012年和2013年分别获fda和ema有条件批准上市,大冢制药的德拉马尼于2013年底获得欧洲ema的有条件批准治疗耐多药结核。
[0008]
普托马尼作用机制是抑制结核杆菌的分枝菌酸的生物合成,从而阻止细胞壁的生成。普托马尼的不良反应:周围神经病变,痤疮,贫血,恶心,呕吐,头痛,转氨酶升高,消化不良,食欲下降,皮疹,瘙痒,腹痛,胸膜炎疼痛,谷氨酰转移酶升高,下呼吸道感染,高淀粉酶血症,咯血,背痛,咳嗽,视力障碍,低血糖,体质量异常减轻和腹泻。
[0009]
2020年01月21日,全球结核病药物开发联盟授予上海复星医药(集团)股份有限公
司全资子公司沈阳红旗制药有限公司在中国境内销售结核病药物普托马尼的许可。
[0010]
在美国和欧盟已经以pretomanid的商品名上市口服固体制剂,规格为200mg。
[0011]
普托马尼的分子结构由4-三氟甲氧基苄基侧链和2-硝基-6-羟基-二氢咪唑并[2, 1-b][1,3]噁嗪核心部分构成,目前后者为结构较为复杂且含有手性碳原子的结构,常见合成的思路为:先构建2-硝基-6-羟基-二氢咪唑并[2,1-b][1,3]噁嗪核心再取代上4-三氟甲氧基苄基侧链,或者顺序相反(参见:tweed c d,dawson r,burger d a,et al. bedaquiline,moxifloxacin,pretomanid,and pyrazinamide during the first 8weeks of treatmentof patients with drug-susceptible or drug-resistant pulmonary tuberculosis:a multicentre,open
‑ꢀ
label,partially randomised,phase 2b trial[j].lancet respir med,2019,7(12):1048-1058.)。而2-硝基-6-羟基-二氢咪唑并[2,1-b][1,3]噁嗪中的手性结构往往通过不对称合成来实现 (参见:rao d r,malhotrag,pullelav s,et al.process for the preparation ofnitroimidazole compounds:in,201621026053[p].2016-07-29.),同时也有手性拆分的方法见诸报道(参见:刘雪英,王力彬,弥乐,等.抗结核候选药物pa-824的合成方法:中国,104177372a[p].2014-12-03)。
[0012]
1997年,pathogenesis公司发明了普托马尼的第一条合成路线,采用硅保护基以及四丁基氟化铵等试剂,价格高昂。2014年上海阳帆公司申请了以2,4-二硝基咪唑为原料的合成线路(申请号201410372418.6),虽然合成路线短,收率高,但使用易爆炸原料2, 4-二硝基咪唑,不利于规模化生产。2021年沈阳药科大学的合成专利(授权公告号:cn 110483549b)获得授权,该方法涉及五步反应,其首先经亲核取代、水解、硅醚化反应得到一个关键中间体6,再经o-烷基化、环合反应得到终产物,虽然整个反应所需原料廉价易得,避免使用易爆炸原料2,4-二硝基咪唑,反应条件温和,但是反应步骤较多,合成路线长,收率不高。
[0013]
综上所述,本发明工艺具有以下优点:
[0014]
1、以2-溴-5-硝基-1h-咪唑为起始原料,避免使用易爆炸原料:2,4-二硝基咪唑,原料均廉价易得,可降低生产成本;
[0015]
2、整个合成线路缩短至三步反应,反应条件温和,总体收率显著提高,安全环保,具有较大优势,适合工业化生产;
[0016]
3、避免使用硅保护基团;通过温和手段(碳酸钾为催化剂)高效构建碳氧键并形成含氧五元杂环。
技术实现要素:
[0017]
针对上述现有技术,本发明的目的是以2-溴-5-硝基-1h-咪唑和缩水甘油丁酸酯为起始原料,在n,n-二异丙基乙胺和甲苯条件下得到(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2
‑ꢀ
羟丙基丁酸酯(中间体ⅰ);用乙腈复溶后,与4-三氟甲氧基溴苄在碳酸铯存在条件下,室温搅拌得到(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-(4-(三氟甲氧基)苄基)氧基) 丁酸丙酯(中间体ⅱ);在0℃时,以甲醇溶解中间体ⅱ,并在碳酸钾条件下搅拌,得到最终产物(s)-6,7-二氢-2-硝基-6-[[4-(三氟甲氧基)苯基]甲氧基]-5h-咪唑并[2,1-b][1, 3]恶嗪,合成方法路线步骤简洁,产物收率高。
[0018]
为实现上述目的,本发明采用如下技术方案:
[0019]
本发明的第一方面,提供一种硝基咪唑吡喃类药物的新合成工艺方法,包括以下步骤:
[0020]
(1)在1000ml得反应瓶中,加入甲苯,2-溴-5-硝基-1h-咪唑,缩水甘油丁酸酯,n, n-二异丙基乙胺。反应完全后将反应液浓缩,残余物经硅胶柱层析,乙酸乙酯石油醚洗脱,得到油状物(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-羟丙基丁酸酯(中间体ⅰ)。合成线路如下:
[0021][0022]
(2)中间体ⅰ溶于乙腈,加入4-三氟甲氧基溴苄、碳酸铯。混合物搅拌过夜。反应完 全后将反应液过滤,滤液减压浓缩。用乙酸乙酯溶解。有机相水洗,饱和氯化钠盐水洗, 无水硫酸钠干燥。干燥的有机相浓缩。残余物过硅胶柱层析,乙酸乙酯石油醚洗脱,得到 (s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-(4-(三氟甲氧基)苄基)氧基)丁酸丙酯 (中间体ⅱ)。
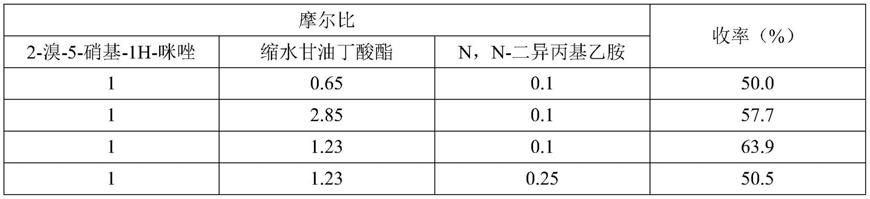
[0023]
(3)中间体ⅱ溶于甲醇,冷却,加入碳酸钾,混合物搅拌,反应完全后过滤,滤液 浓缩。残余物过硅胶柱层析,乙酸乙酯石油醚洗脱,得到最终产物(s)-6,7-二氢-2-硝 基-6-[[4-(三氟甲氧基)苯基]甲氧基]-5h-咪唑并[2,1-b][1,3]恶嗪。
[0024]
步骤(1)中,所述优选的2-溴-5-硝基-1h-咪唑:缩水甘油丁酸酯:n,n-二异丙基乙胺的物料摩尔比例为1:1.23:0.10。
[0025]
步骤(1)中,优选的混合物反应温度为75℃,反应时间为6h。
[0026]
步骤(1)中,优选的乙酸乙酯和石油醚洗脱比例为2:3。
[0027]
步骤(1)中,优选的硅胶层析柱垫料目数为200目。
[0028]
步骤(2)中,优选的中间体ⅰ:4-三氟甲氧基溴苄:碳酸铯的摩尔比例为1:1.05:2.44。
[0029]
步骤(2)中,优选的乙酸乙酯和石油醚洗脱比例为1:1。
[0030]
步骤(2)中,优选的混合物反应温度为25℃,反应时间为14h。
[0031]
步骤(2)中,优选的硅胶层析柱垫料目数为300目。
[0032]
步骤(3)中,优选的中间体ⅱ:碳酸钾的物料摩尔比例为1:3.02。
[0033]
步骤(3)中,优选的中间体ⅱ溶解于极性溶剂后的冷却温度为0℃。
[0034]
步骤(3)中,优选的乙酸乙酯和石油醚洗脱比例为2:1。
[0035]
步骤(3)中,优选的混合物反应温度为10℃,反应时间为2.5h。
[0036]
步骤(3)中,优选的硅胶层析柱垫料目数为300目。
[0037]
步骤(1)(2)(3)中,溶解中间体ⅰ优选乙腈、溶解中间体ⅱ优选甲醇,洗脱时极性溶剂优选乙酸乙酯,非极性溶剂优选石油醚。
[0038]
本发明的第二方面,提供上述方法制备的原料药的安全性证明。
[0039]
本发明的有益效果:
[0040]
采用本发明的一种硝基咪唑吡喃类药物合成方法以2-溴-5-硝基-1h-咪唑为起始原料,避免使用易爆炸原料:2,4-二硝基咪唑,原料均廉价易得,可降低生产成本;整个合成线路缩短至三步反应,反应条件温和,总体收率显著提高,安全环保,具有较大优势,适合工
业化生产;避免使用硅保护基团;通过温和手段(碳酸钾为催化剂)高效构建碳氧键并形成含氧五元杂环。
具体实施方式
[0041]
应该指出,以下详细说明都是例示性的,旨在对本技术提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本技术所属技术领域的普通技术人员通常理解的相同含义。
[0042]
正如背景技术部分介绍的,本发明缩短合成路线,避免使用高昂和易燃易爆的试剂,适合工业化生产。
[0043]
为了使得本领域技术人员能够更加清楚地了解本技术的技术方案,以下将结合具体的实施例详细说明本技术的技术方案。
[0044]
本发明实施例中所用的未进行具体说明试验材料均为本领域常规的试验材料,均可通过商业渠道购买得到。
[0045]
实施例1:中间体ⅰ的制备(不同摩尔比对于产物收率)
[0046]
合成线路为:在1000ml得反应瓶中,加入甲苯,2-溴-5-硝基-1h-咪唑,缩水甘油丁酸酯,n,n-二异丙基乙胺。混合物反应温度为75℃,反应时间为6h,反应完全后将反应液浓缩,残余物经200目硅胶柱层析,乙酸乙酯石油醚(比例为2:3)洗脱,得到油状物(s)
ꢀ‑
3-(2-溴-4-硝基-1h-咪唑-1-基)-2-羟丙基丁酸酯(中间体ⅰ)。
[0047]
按照如上反应方程式进行反应,结果如下:
[0048][0049]
实施例2:中间体ⅰ的制备(不同反应温度、反应时间、硅胶目数、洗脱液比例对于产物收率)
[0050]
合成线路为:在1000ml得反应瓶中,加入250ml甲苯,0.3mol 2-溴-5-硝基-1h-咪唑, 0.37mol缩水甘油丁酸酯,32mmol n,n-二异丙基乙胺。反应完全后将反应液浓缩,残余物经硅胶柱层析,乙酸乙酯石油醚洗脱,得到油状物(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)
ꢀ‑
2-羟丙基丁酸酯(中间体ⅰ)。
[0051]
按照如上反应方程式进行反应,结果如下:
[0052]
反应温度(℃)反应时间(h)硅胶目数乙酸乙酯石油醚比例收率(%)4562002:351.145122002:353.04583001:154.27562002:363.97563002:360.77562001:157.4
10532002:355.010562001:150.2
[0053]
实施例3:中间体ⅱ的制备(不同摩尔比对于产物收率)
[0054]
合成线路为:中间体ⅰ溶于乙腈,加入4-三氟甲氧基溴苄、碳酸铯。混合物搅拌,反应温度为25℃,反应时间为14h。反应完全后将反应液过滤,滤液减压浓缩。用乙酸乙酯溶解。有机相水洗,饱和氯化钠盐水洗,无水硫酸钠干燥。干燥的有机相浓缩。残余物过 300目硅胶柱层析,乙酸乙酯石油醚(比例为1:1)洗脱,得到(s)-3-(2-溴-4-硝基-1h
‑ꢀ
咪唑-1-基)-2-(4-(三氟甲氧基)苄基)氧基)丁酸丙酯(中间体ⅱ)。
[0055]
按照如上反应方程式进行反应,结果如下:
[0056][0057]
实施例4:中间体ⅱ的制备(不同反应温度、反应时间、硅胶目数、洗脱液比例对于产物收率)
[0058]
合成线路为:94.3mmol中间体ⅰ溶于190ml乙腈,加入95.4mmol 4-三氟甲氧基溴苄、 0.23mol碳酸铯。混合物搅拌,反应完全后将反应液过滤,滤液减压浓缩。用乙酸乙酯溶解。有机相水洗,饱和氯化钠盐水洗,无水硫酸钠干燥。干燥的有机相浓缩。残余物过硅胶柱层析,乙酸乙酯石油醚洗脱,得到(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-(4-(三氟甲氧基)苄基)氧基)丁酸丙酯(中间体ⅱ)。
[0059]
按照如上反应方程式进行反应,结果如下:
[0060]
反应温度(℃)反应时间(h)硅胶目数乙酸乙酯石油醚比例收率(%)10143002:150.010204001:153.21083001:147.725143001:158.82584002:156.340144001:155.24083001:256.340203001:145.9
[0061]
实施例5:最终产物的制备(不同摩尔比对于产物收率)
[0062]
合成线路为:中间体ⅱ溶于甲醇,冷却至0℃,加入碳酸钾(或其他),混合物搅拌,反应温度为10℃,反应时间为2.5h,反应完全后过滤,滤液浓缩。残余物过300目硅胶柱层析,乙酸乙酯石油醚(比例为2:1)洗脱,得到最终产物(s)-6,7-二氢-2-硝基-6-[[4
‑ꢀ
(三氟甲氧基)苯基]甲氧基]-5h-咪唑并[2,1-b][1,3]恶嗪。
[0063]
按照如上反应方程式进行反应,结果如下:
[0064][0065]
实施例6:最终产物的制备(不同反应温度、反应时间、硅胶目数、洗脱液比例对于产物收率)
[0066]
86mmol中间体ⅱ溶于200ml甲醇,冷却,加入260mmol碳酸钾,混合物搅拌,反应完全后过滤,滤液浓缩。残余物过硅胶柱层析,乙酸乙酯石油醚洗脱,得到最终产物(s)
ꢀ‑
6,7-二氢-2-硝基-6-[[4-(三氟甲氧基)苯基]甲氧基]-5h-咪唑并[2,1-b][1,3]恶嗪。
[0067]
按照如上反应方程式进行反应,结果如下:
[0068][0069]
实施例7:合成案例
[0070]
(1)在1000ml得反应瓶中,加入150ml甲苯,0.2mol 2-溴-5-硝基-1h-咪唑,0.25 mol缩水甘油丁酸酯,0.03mol n,n-二异丙基乙胺。混合物反应温度为55℃,反应时间为8h,反应完全后将反应液浓缩,残余物经200目硅胶柱层析,乙酸乙酯石油醚(比例为1:1)洗脱,得到油状物(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-羟丙基丁酸酯(中间体ⅰ)。
[0071]
(2)56.2mmol中间体ⅰ溶于100ml乙腈,加入63.1mmol 4-三氟甲氧基溴苄、0.17 mol碳酸铯。混合物搅拌,反应温度为10℃,反应时间为20h。反应完全后将反应液过滤,滤液减压浓缩。用乙酸乙酯溶解。有机相水洗,饱和氯化钠盐水洗,无水硫酸钠干燥。干燥的有机相浓缩。残余物过300目硅胶柱层析,乙酸乙酯石油醚(比例为1:1)洗脱,得到(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-(4-(三氟甲氧基)苄基)氧基)丁酸丙酯(中间体ⅱ)。
[0072]
(3)98.4mmol中间体ⅱ溶于300ml甲醇,冷却,加入278.4mmol碳酸钾,混合物搅拌,反应温度为15℃,反应时间为2h,反应完全后过滤,滤液浓缩。残余物过300目硅胶柱层析,乙酸乙酯石油醚(比例为1:1)洗脱,得到最终产物(s)-6,7-二氢-2-硝基-6-[[4
‑ꢀ
(三氟甲氧基)苯基]甲氧基]-5h-咪唑并[2,1-b][1,3]恶嗪。实得最终产物收率为:57.3%,纯度大
于99%(hplc检测),色谱条件为:色谱柱:diamonsil c18柱(250mm
×
4.6mm,5μm)、检测波长:321nm、流动相:乙腈:水-30:70(v:v)、流速:1.0ml/min、柱温: 85℃
±
5℃。
[0073]1h nmr(400mhz,cdcl3):δ7.99(s,1h),δ7.39(d,j=8.8hz,2h),δ7.36 (d,j=8.5hz,2h),δ4.65-4.72(m,3h),4.50(d,j=11.5hz,1h),4.23-4.40(m, 3h)。
[0074]
13
c nmr(100mhz,cdcl3):δ149.73,147.65,143.34,135.01,129.40,121.36, 120.14,116.79,111.01,69.56,67.26,66.08,46.95。
[0075]
hrms(esi-ms)calcd.for c
15h14
f3n2o5,[m h] 360.54,[m na] 382.81。
[0076]
实施例8:合成案例
[0077]
(1)在1000ml得反应瓶中,加入650ml甲苯,0.45mol 2-溴-5-硝基-1h-咪唑,0.78 mol缩水甘油丁酸酯,0.09mol n,n-二异丙基乙胺。混合物反应温度为75℃,反应时间为6h,反应完全后将反应液浓缩,残余物经200目硅胶柱层析,乙酸乙酯石油醚(比例为2:3)洗脱,得到油状物(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-羟丙基丁酸酯(中间体ⅰ)。
[0078]
(2)107.6mmol中间体ⅰ溶于210ml乙醚,加入105.4mmol 4-三氟甲氧基溴苄、 0.41mol碳酸铯。混合物搅拌,反应温度为25℃,反应时间为14h。反应完全后将反应液过滤,滤液减压浓缩。用乙酸乙酯溶解。有机相水洗,饱和氯化钠盐水洗,无水硫酸钠干燥。干燥的有机相浓缩。残余物过300目硅胶柱层析,乙酸乙酯石油醚(比例为1:1)洗脱,得到(s)-3-(2-溴-4-硝基-1h-咪唑-1-基)-2-(4-(三氟甲氧基)苄基)氧基)丁酸丙酯(中间体ⅱ)。
[0079]
(3)150mmol中间体ⅱ溶于300ml异丙醇,冷却,加入600mmol氢氧化钠,混合物搅拌,反应温度为10℃,反应时间为2.5h,反应完全后过滤,滤液浓缩。残余物过400目硅胶柱层析,乙酸乙酯石油醚(比例为2:1)洗脱,得到最终产物(s)-6,7-二氢-2-硝基
ꢀ‑
6-[[4-(三氟甲氧基)苯基]甲氧基]-5h-咪唑并[2,1-b][1,3]恶嗪。实得最终产物收率为: 59.8%,纯度大于99%(hplc检测),色谱条件为:色谱柱:diamonsil c18柱(250mm
×ꢀ
4.6mm,5μm)、检测波长:321nm、流动相:乙腈:水-30:70(v:v)、流速:1.0ml/min、柱温:85℃
±
5℃。
[0080]1h nmr(400mhz,cdcl3):δ7.84(s,1h),δ7.36(d,j=8.5hz,2h),δ7.25 (d,j=7.5hz,2h),δ4.75(d,j=7.8hz,1h),δ4.71-4.73(m,2h),δ4.41(d,j=11.8hz,1h),δ4.10-4.18(m,3h)。
[0081]
13
c nmr(100mhz,cdcl3):δ150.12,147.65,143.34,135.01,128.93,121.43, 119.04,115.19,70.03,67.32,65.45,47.34。
[0082]
hrms(esi-ms)calcd.for c
15h14
f3n2o5,[m h] 360.37,[m na] 382.59。
[0083]
实施例9:化合物急性毒性研究
[0084]
取实施实例7合成的化合物,研究在icr小鼠体内的安全性。化合物以可食用植物油溶解。
[0085]
种属和品系:icr小鼠;等级:spf级;数量和性别:40只,雌雄各半;开始给药时年龄范围:约4~5周;开始给药时体重范围:18-24g;实验动物分组方法:雌雄动物分别按体重进行分层随机分组,分为阴性对照组、给药组,20只/组,雌雄各半,给药前禁食过夜。按照2000mg/kg的剂量对给药组进行口服灌喂,阴性对照组灌喂等量可食用植物油,试验后小鼠出现均未出现异常活动,未出现死亡个例,所用动物继续饲养14天,本试验全过程中没有死亡或濒死动物,所有存活动物于第15天进行了大体解剖,动物体表及各主要器官大体检查均未发现明显形态学异常。说明合成的化合物安全性较好。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。