1.本发明涉及海洋真菌活性成分分析技术领域,具体涉及一种吲哚生物碱化合物及其制备方法和应用。
背景技术:
2.海洋微生物是海洋中一种重要的生物资源,微生物参与海洋物质分解和转化的全过程,具有复杂而重要的生态功能。海洋微生物能够耐受海洋特有的如高盐、高压、低氧、低光照等极端条件,生活环境的特异性导致海洋微生物在物种、基因组成和生态功能上的多样性。
3.海洋微生物独特的生存环境使其具有产生新型生物活性物质的巨大潜力,从海洋微生物次级代谢产物中筛选药用活性物质成为当前研究热点。海洋微生物中的海洋真菌是活性次级代谢产物的丰富来源,其具有生命力强,代谢旺盛等特点,而且海洋真菌的次级代谢产物70-80%具有生物活性,以海洋真菌为原料发现具有特定结构类型的天然产物对于海洋药物的研发具有重要意义。
4.曲霉属真菌土曲霉(aspergillus cf.terreus)属于半知菌纲,壳霉目,杯霉科。目前针对土曲霉次级代谢产物的研究,主要成果集中于聚酮类、萜类和油脂类化合物等,具有丰富且潜力十足的生物活性,如抗炎、抗菌以及抗肿瘤等。例如,雷丹丹等从内生菌aspergillus terreus pc-038次生代谢产物分离到化合物1,2-dihydrohelvolic acid、烟曲霉酸对金黄色葡萄球菌 (staphylococcus aureus)atcc 25923有显著的抑制作用,为新型抗生素的研发奠定了基础[内生菌aspergillus terreus pc-038次生代谢产物中抗菌化学成分研究,化学试剂,2021,43(08),1143-1150.]。
[0005]
陈修文等从深海来源曲霉16-02-1的代谢产物中分离到新曲霉酸、 ferrineoaspergillin、(2’s)-4-甲氧基-3-(2
’‑
甲基-3’羟基)丙羟基-苯甲酸甲酯、黄曲霉素等化合物,对人慢性粒细胞白血病k562细胞、人早幼粒白血病 hl-60细胞、人宫颈癌hela细胞、人胃腺癌bgc-823细胞有一定抑制作用[深海来源曲霉16-02-1的代谢产物及其抗肿瘤抗真菌活性初步测评,中国海洋药物,2013年6月,第32卷第3期]。
[0006]
近年来从土曲霉代谢产物中分离并报道的化合物类型多样且具有较好的药理活性,但是迄今还未查到有文献报道从土曲霉代谢产物中分离得到具有抗肿瘤活性的吲哚生物碱化合物。
技术实现要素:
[0007]
本发明的目的在于从海洋土曲霉菌的代谢产物中提取获得具有药用价值的天然活性物质。
[0008]
为实现上述目的,本发明采用如下技术方案:
[0009]
本发明从中国 台湾海洋沉积物中分离得到曲霉属真菌土曲霉(aspergillus cf.terreus)cxx-158-20,对其在大米固体培养基中培养产生的次级代谢产物的化学成分
及其生物活性进行了深入的研究,从中分离纯化得到4个新的单体吲哚生物碱化合物,所述化合物的结构式选自式(ⅰ)-(ⅳ),
[0010][0011]
上述4个化合物为具有吲哚和多取代苯环的吲哚生物碱化合物,结构式如式(ⅰ)所示的土曲吲哚宁a(化合物i)的分子式为c
24h20
n2o5,结构式如式(ⅱ)所示的土曲吲哚宁b(化合物ii)的分子式为c
22h19
no6,结构式如式(ⅲ)所示的土曲吲哚宁c(化合物iii)的分子式为c
25h20
n2o6,结构式如式(ⅳ)所示的土曲吲哚宁d(化合物vi)的分子式为c
26h28
n2o6。
[0012]
本发明还提供了所述吲哚生物碱化合物的制备方法,包括以下步骤:
[0013]
(1)将保藏号为cctcc no:m 20211214的曲霉属真菌(aspergillus cf. terreus)cxx-158-20活化后接种于大米固体培养基中,20-30℃静态培养 10-40天;
[0014]
(2)发酵培养结束后,分离得到菌丝体和发酵液;
[0015]
(3)将菌丝体加入甲醇中浸提,分离得到浸提液,浸提液浓缩后用蒸馏水混悬,得到水混悬液,然后用乙酸乙酯对水混悬液进行萃取,萃取液分离纯化后,制得吲哚生物碱化合物;
[0016]
利用乙酸乙酯对发酵液进行萃取,萃取液分离纯化制得吲哚生物碱化合物;
[0017]
所述分离纯化包括:萃取液利用正相硅胶柱层析,依次以体积比为 100:0,98:2,95:5,90:10,80:20,70:30,40:60,0:100的石油醚/乙酸乙酯混合液梯度洗脱,收集80:20馏分利用甲醇重结晶得到化合物i;收集 70:30馏分用体积比3:1的甲醇/乙酸乙酯混合液重结晶得到化合物ii;
[0018]
萃取液利用反相硅胶柱层析,以体积比为30%~100%的甲醇/水溶液梯度洗脱,收集85%~90%馏分得到化合物iii;收集90%~95%馏分得到化合物vi;
[0019]
或者,将萃取液进行正相硅胶柱层析,依次以体积比9:1、8:1、7:1、 6:1、5:1、4:1、3:1、2:1、1:1、1:3、1:9的石油醚/乙酸乙酯混合液和乙酸乙酯进行梯度洗脱,收集体积比4:1的石油醚/乙酸乙酯混合液洗脱出的子馏分a和3:1的子馏分b;子馏分a通过制备型高效液相色谱层析得到化合物i和化合物ii;子馏分b先利用反相硅胶柱层析,再经制备型高效液相色谱层析得到iii和化合物vi。
[0020]
步骤(1)中,对土曲霉aspergillus cf.terreus cxx-158-20进行发酵培养。
[0021]
作为优选,菌种活化采用pda培养基,以体积1l计,包括以下原料:土豆200g、葡萄糖20g、琼脂20g、余量为h2o,ph自然。
[0022]
发酵培养采用大米固体培养基,具体的,包括以下原料:每50g大米中添加75ml水,ph为自然。
[0023]
优选的,发酵培养的温度为22-26℃。更优选为25℃培养20天。
[0024]
步骤(2)中,分离获得菌丝体和发酵液,所述的菌丝体和发酵液中均能提取分离得到吲哚生物碱化合物。
[0025]
其中,利用菌丝体获取吲哚生物碱化合物时,将菌丝体置于甲醇中浸泡7-14天,菌丝体充分破壁,使胞内物质有效溶出。再利用乙酸乙酯对目标产物进行萃取。
[0026]
利用发酵液获取吲哚生物碱化合物时,先将发酵液与硅藻土搅拌后,采用乙酸乙酯回流进行萃取。
[0027]
利用正相硅胶柱层析、反相硅胶柱层析、重结晶、高效液相色谱分离等技术手段对目标产物进行分离纯化。通过多步分离纯化,能获得纯度较高的生物碱化合物。
[0028]
具体的,萃取液经正相硅胶柱层析后收集的子馏分a,再进行高效液相色谱分离,流动相采用63%乙腈-水,流速3ml/min,收集保留时间 17.8min的馏分得到化合物i;收集保留时间19.1min的馏分得到化合物ii;
[0029]
子馏分b经ods开放柱层析,以甲醇-水(30%~100%)梯度洗脱,收集 60%~100%洗脱的子馏分fr.3,再进行高效液相色谱分离,流动相采用68%甲醇-水,流速3ml/min,收集保留时间13.0min的馏分得到化合物iii;收集保留时间16.6min的馏分得到化合物vi。
[0030]
本发明研究表明,利用上述方法从发酵培养物中分离得到的吲哚生物碱化合物具有较好的抗肿瘤活性,针对肿瘤细胞hela,化合物i的ic
50
值为10.3μm,化合物ii的ic
50
值为13.5μm,化合物iii的ic
50
值为34.8μm,化合物iv的ic
50
值为43.7μm;针对肿瘤细胞a549,化合物i的ic
50
值为32.8μm,化合物ii的ic
50
值为24.9μm,化合物iii的ic
50
值为41.0μm,化合物iv的ic
50
值为25.7μm。
[0031]
因此,本发明还提供了所述的吲哚生物碱化合物在制备抗肿瘤药物中的应用。
[0032]
进一步的,所述肿瘤为宫颈癌或肺癌。
[0033]
本发明还提供了一种药物组合物,包括有效剂量的吲哚生物碱化合物和药学上可接受的载体。
[0034]
所述药物组合物以本发明的吲哚生物碱化合物为主要活性成分,添加药剂学上可接受的辅料制成,可按照药剂学上记载的制剂制备方法制成制剂。所述的制剂形式可以为
注射液、滴注液、粉针剂、颗粒剂、片剂、冲剂、散剂、口服液、糖衣片剂、薄膜衣片剂、肠溶衣片剂、口含剂、颗粒剂、丸剂、膏剂、丹剂、喷雾剂、滴丸剂、崩解剂、口崩片、微丸等。
[0035]
本发明具备的有益效果:
[0036]
(1)本发明从海洋真菌的发酵培养物中提取、分离获得了一种具有新颖结构的吲哚生物碱化合物,该方法操作简便、提取得率高、产物纯度高,适合规模化生产。
[0037]
(2)通过体外抗肿瘤试验,表明本发明提供的吲哚生物碱化合物具有较好的抗肿瘤活性,可将其用于制备抗肿瘤药物,具有良好的开发前景。
附图说明
[0038]
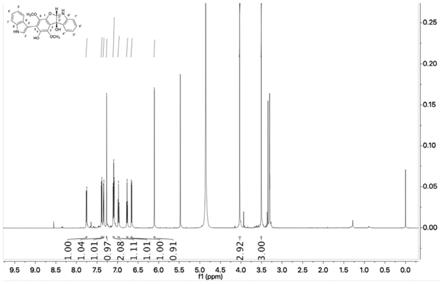
图1为本发明化合物i的1h nmr图谱(in cd3od)。
[0039]
图2为本发明化合物i的
13
c nmr图谱(in cd3od)。
[0040]
图3为本发明化合物ii的1h nmr图谱(in cd3od)。
[0041]
图4为本发明化合物ii的
13
c nmr图谱(in cd3od)。
[0042]
图5为本发明化合物iii的1h nmr图谱(in cd3od)。
[0043]
图6为本发明化合物iii的
13
c nmr图谱(in cd3od)。
[0044]
图7为本发明化合物iv的1h nmr图谱(in cd3od)。
[0045]
图8为本发明化合物iv的
13
c nmr图谱(in cd3od)。
具体实施方式
[0046]
下面结合具体实施例对本发明做进一步说明。以下实施例仅用于说明本发明,不用来限制本发明的适用范围。在不背离本发明精神和本质的情况下,对本发明方法、步骤或条件所做的修改或替换,均属于本发明的范围。
[0047]
下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
[0048]
实施例中采用的真菌土曲霉(aspergillus cf.terreus)cxx-158-20分离自中国 台湾海洋沉积物,经分子生物学与菌落形态鉴定为曲霉属真菌土曲霉。于2021年9月24日保藏于中国典型培养物保藏中心,保藏地址:中国,武汉,武汉大学,保藏号为:cctcc no:m 20211214,于2021年10月 9日检测为存活。
[0049]
pda固体培养基的制备:土豆200g、葡萄糖20g、琼脂20g、dd h2o 补齐1l,121℃灭菌30min,ph自然。
[0050]
大米固体培养基的制备:每50g大米添加75mldd h2o,121℃灭菌 30min,ph自然。
[0051]
实施例1真菌土曲霉aspergillus cf.terreus cxx-158-20的分离
[0052]
先将海底沉积物样品放在无菌的培养皿中,在自然环境中风干,然后称取1g风干沉积物样品,用10ml 50%的海水溶解得10-1
溶液,继续稀释到10-2
、10-3
,得三个梯度的样品液。分别取100μl涂布在分离培养基gpy 培养基、martin培养基、ca培养基、pda培养基、sda培养基及察氏培养基上,每个不同稀释浓度的分离培养基2个平行,在28℃下培养1~8周。观察并挑取其中的单菌落,接种在察氏培养基上,获得纯培养菌株,经分子生物学与菌落形态鉴定为曲霉属真菌土曲霉,命名为aspergillus cf. terreus cxx-158-20。
[0053]
实施例2真菌土曲霉aspergillus cf.terreus cxx-158-20的发酵培养
[0054]
将真菌土曲霉aspergillus cf.terreus cxx-158-20制成孢子悬浮液,接种到pda固体培养基中活化,再接种到在大米固体培养基中进行大量发酵,在室温下静置发酵20天。菌丝体用甲醇过夜浸泡菌丝体进行提取。
[0055]
实施例3生物碱化合物的制备
[0056]
真菌土曲霉aspergillus cf.terreus cxx-158-20发酵培养后,取5l发酵培养液,离心取沉淀得到菌丝体;菌丝体用甲醇浸泡1周,浸泡液经浓缩后用1l蒸馏水混悬,水混悬液合并,用6l乙酸乙酯萃取,乙酸乙酯萃取液经浓缩得到浸膏10g;用正相硅胶(100目,100g)拌样,提取物经硅胶开放柱层析(φ50
×
640mm,200~300目,500~700g),以石油醚-乙酸乙酯 (100:0,98:2,95:5,90:10,80:20,70:30,40:60,0:100)和乙酸乙酯-甲醇 (95:5、90:10)梯度洗脱,每次300ml;tlc检测馏分。收集石油醚-乙酸乙酯80:20馏分再用甲醇(室温,100ml)重结晶得到主要为化合物i的粗结晶a。粗结晶a再用甲醇(室温,30ml)重结晶得到纯化合物i。收集石油醚
ꢀ‑
乙酸乙酯70:30馏分用甲醇:乙酸乙酯(3:1)重结晶得到主要为化合物ii 的粗结晶b。粗结晶b再用甲醇(室温,30ml)重结晶得到纯化合物ii。
[0057]
母液经反相硅胶ods开放柱层析以甲醇/水(30%~100%)梯度洗脱。合并馏分85%和90%(甲醇/水)得到化合物iii的粗品,用甲醇重结晶得到化合物iii的纯品。合并馏分90%和95%(甲醇/水)得到化合物iv的粗品,用甲醇重结晶得到化合物iv的纯品。
[0058]
实施例4生物碱化合物的制备
[0059]
真菌土曲霉aspergillus cf.terreus cxx-158-20发酵培养后,取5l发酵培养液,离心取上清得到发酵液;发酵液浓缩,用10g硅藻土拌样,1l 乙酸乙酯回流,进行正相硅胶柱层析分离(200-300目,1kg;硅胶柱尺寸l 50mm,),依次以体积比9:1、8:1、7:1、6:1、5:1、4:1、3:1、2:1、 1:1、1:3、1:9的石油醚/乙酸乙酯混合液和乙酸乙酯进行梯度洗脱,收集体积比4:1的石油醚/乙酸乙酯混合液洗脱出的子馏分a和3:1的子馏分b。
[0060]
子馏分a通过制备型hplc(ymc-pack ods-a,63%乙腈-水,3ml/min) 得化合物i(保留时间rt:17.8min)和化合物ii(保留时间rt:19.1min)。
[0061]
子馏分b经ods开放柱层析,以甲醇/水(30%~100%)梯度洗脱,共得到3个子馏分:fr.1(甲醇/水(30%~40%))、fr.2(甲醇/水(40%~60%))、fr.3(甲醇/水(60%~100%))。子馏分fr.3通过制备型hplc(ymc-pack ods-a,68%甲醇/水,3ml/min)得到化合物iii(保留时间rt:13.0min)和化合物vi(保留时间rt:16.6min)。
[0062]
实施例5生物碱化合物i的结构鉴定
[0063]
采用hplc对制得的化合物进行纯度鉴定,纯度大于98%的样品运用质谱和核磁共振技术进行结构鉴定,核磁共振用日本电子公司jeol600mhz nmr sectrometer测定,tms作内标;高分辨质谱用美国agilent 公司6230tof lc/ms spectrometer测定。
[0064]
化合物i为黑色固体粉末,分子旋光 88.00(c 0.1,meoh)。其高分辨质谱(hresims)测定结果为m/z 417.1459[m h]
,结合
13
c-nmr 数据推测其分子式为c
24h20
n2o5,不饱和度为16。
[0065]
通过进一步分析该化合物的氢谱、碳谱(图1、图2、表1)和hsqc,判断该结构中可能含有2个甲氧基[δ
h 3.5(s,och
3-6,δ
c 60.6)]和[δ
h 4.0(s, och
3-3,δ
c 62.1)];8个芳香h,分别是[δ
h 7.75(d,j=8.5hz,h-4”,δ
c 126.6), δ
h 6.77(t,j=7.9hz,h-5”,δ
c 120.1),δ
h 7.11
–
7.07(m,h-6”,δ
c 130.6),δ
h 6.66(d,j=7.9hz,h-7”,δ
c 110.6),δ
h 7.34(d,j=
8.0hz,h-4’,δ
c 121.3), δ
h 6.98(t,j=8.0hz,h-5’,δ
c 120.1),δ
h 7.11
–
7.07(m,h-6’,δ
c 122.4),δ
h 7.39(d,j=8.1hz,h-7’,δ
c 112.3)];2个sp 3
杂化的次甲基[δ
h 7.27(s,h-2’, δ
c 126.4),δ
h 6.11(s,h-2”,δ
c 107.0)]。
[0066]
根据1h-1
h cosy相关和hmbc信号,将[δ
c 126.6(c-4”),120.1 (c-5”),130.6(c-6”),110.6(c-7”),150.8(c-8”),131.3(c-9”)]分配给一个苯环,[δ
c 121.3(c-4’),120.1(c-5’),122.4(c-6’),112.3(c-7’),137.9 (c-8’),128.6(c-9’)]分配给另一个苯环,同时确定了c4
”‑
c5”,c5
”‑
c6”, c6
”‑
c7”,c4
’‑
c5’,c6
’‑
c7’相关片段,以及这两个苯环均为1,2-二取代苯环。六个共振的[δ
c 146.4(c-1),123.4(c-2),142.0(c-3),143.6(c-4),119.7 (c-5),139.8(c-6)]构成了第三个六取代苯环。
[0067]
hmbc显示δ
h 6.11(s,h-2”,δ
c 107.0)与δ
c 150.8(c-8”),131.3(c-9”), 146.4(c-1)有相关信号,以此确定二氢呋喃环与二氢吡咯烷环和六取代苯环的链接。
[0068]
noesy信号显示,δ
h 4.0(s,och
3-3)与δ
h 7.75(d,j=8.5hz,h-4”) 有相关,δ
h 3.5(s,och
3-6)与δ
h 7.27(s,h-2’)有相关,且δ
h 4.0(s, och
3-4)与δ
h 3.5(s,och
3-3)无相关信号,确定两个甲氧基分别位于c-3 和c-6上,而oh位于c-4上。
[0069]
表1生物碱化合物i的nmr数据
[0070]
4150.2,c-5126.1,c-6135.1,c-2”107.3,ch6.11,s3”90.7,c-4”126.8,ch7.84,d(7.4)5”120.1,ch6.74,t(7.5)6”130.5,ch7.08,t(7.7)7”110.5,ch6.65,d(7.9)8”150.7,c-9”131.4,c-1’131.2,c-2’115.7,ch6.80,d(8.5)3’132.7,ch7.18,d(8.5)4’157.8,c-5’132.7,ch7.18,d(8.5)6’115.7,ch6.80,d(8.5)c
3-och361.0,ch33.23,sc
6-och360.9,ch33.50,s
[0080]
由此确定了化合物ii的结构如下所示:
[0081][0082]
实施例7生物碱化合物iii的结构鉴定
[0083]
采用hplc对制得的化合物进行纯度鉴定,纯度大于98%的样品运用质谱和核磁共振技术进行结构鉴定,核磁共振用日本电子公司jeol 600mhz nmr sectrometer测定,tms作内标;高分辨质谱用美国agilent 公司6230 tof lc/ms spectrometer测定。
[0084]
化合物iii为棕色固体粉末,分子旋光 82.00(c 0.1,meoh)。其高分辨质谱(hresims)测定结果为m/z 445.1403[m h]
,结合
13
c-nmr 数据推测其分子式为c
25h20
n2o6,不饱和度为17。
[0085]
化合物iii的核磁共振数据(图5、图6、表3)显示了与化合物i相似的结构特征,该化合物的氢谱、碳谱和hsqc表明其较化合物i多了一个次甲基δ
c 163.1(c-8”),δ
h 8.81(s,h-8”),hmbc信号显示,δ
h 8.81(s,h-8”) 与δ
c 141.6(c-9”)存在明显相关,而且δ
h 6.46(s,h-2”)与δ
c 163.1(c-8”)存在相关信号。同时根据高分辨质谱结果显示的分子质量和化学位移变化我们认为是1-nh变成了1-noh。
[0086]
表3生物碱化合物iii的nmr数据
[0087][0088][0089]
由此我们确定了化合物iii的结构如下所示:
[0090][0091]
实施例8生物碱化合物iv的结构鉴定
[0092]
采用hplc对制得的化合物进行纯度鉴定,纯度大于98%的样品运用质谱和核磁共振技术进行结构鉴定,核磁共振用日本电子公司jeol600mhz nmr sectrometer测定,tms作内标;高分辨质谱用美国agilent 公司6230 tof lc/ms spectrometer测定。
[0093]
化合物iv为为棕色油状液体。其高分辨质谱(hresims)测定结果为m/z 467.1968[m h]
,结合
13
c-nmr数据推测其分子式为c
26h28
n2o6,不饱和度为14。
[0094]
通过进一步分析该化合物的nmr谱图(图7、图8、表4),发现其与化合物i具有相似
的骨架,通过与化合物i的对比,我们发现化合物iv的 h-2’,h-2”,h-3”消失取而代之的是c2
”‑
c3”的双键和c-2’的异戊二烯基。根据h-14’(δ
h 1.36,s,3h)和c-2’(δ
c 141.7),c-11’(δ
c 146.9)存在bc相关,h-11’(δ
h 6.11,dd,j=17.4,10.6hz,1h)和c-2’(δ
c 141.7),c-13’(δ
c 26.8)存在bc相关,同时h-11’/h-12’存在1h-1
h cosy自旋耦合,验证了异戊二烯基的存在且取代在c-2’。
[0095]
表4生物碱化合物iv的nmr数据
[0096][0097]
[0098]
由此我们确定了化合物iv的结构如下所示:
[0099][0100]
实施例9生物碱化合物抗肿瘤活性分析
[0101]
hela和a549细胞培养于含10%小牛血清、青霉素100iu/ml及链霉素100g/ml的rp-mi 1640培养基中,每3d换液1次,每5d传代1次。细胞均置于37℃。取对数生长期细胞,以rpmi 1640培养基稀释成 5
×
104/ml单细胞悬液,接种于96孔细胞培养板,每个浓度复种3孔,每孔180μl。置培养箱温育12h后,药物组每孔加不同浓度供试液20μl,平行设空白对照组(用等体积的rpmi 1640培养基代替受试药物),阳性对照组(5-fu)共培养48h。每孔加入1mg/ml mtt溶液50μl,继续培养4h 后,吸尽上清液,每孔加入二甲基亚砜(dmso)150μl,充分溶解mtt还原产物。置酶标仪上于492nm波长处测定各药物组和空白组的光密度(d),按公式计算得到药物对肿瘤细胞的半数抑制浓度(ic
50
),并对药效进行初步的评价。
[0102]
ir(%)=(1-加药组平均d值/对照组平均d值)
×
100%。
[0103]
实验结果显示,4种生物碱化合物具有较好的抗肿瘤活性,针对肿瘤细胞hela,化合物i的ic
50
值为10.3μm,化合物ii的ic
50
值为13.5μm,化合物iii的ic
50
值为34.8μm,化合物iv的ic
50
值为43.7μm,阳性对照5-fu的ic
50
值为18.3μm;针对肿瘤细胞a549,化合物i的ic
50
值为 32.8μm,化合物ii的ic
50
值为24.9μm,化合物iii的ic
50
值为41.0μm,化合物iv的ic
50
值为25.7μm,阳性对照5-fu的ic
50
值为24.2μm。表明该化合物具有较好的抗肿瘤作用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。